
|
Filed pursuant to Rule 424(b)(3) Registration No. 333-249539 |
PROSPECTUS SUPPLEMENT NO. 1
(to Prospectus dated August 24, 2021)
180 LIFE SCIENCES CORP.
Common Stock
This prospectus supplement updates, amends, and supplements the prospectus dated August 24, 2021 (the “Prospectus”), which forms a part of our Registration Statement on Form S-1 (Registration No. 333-249539).
This prospectus supplement is being filed to update, amend, and supplement the information in the Prospectus with the information contained in our Annual Report on Form 10-K, filed with the Securities and Exchange Commission on March 31, 2022 (the “Annual Report”). Accordingly, we have attached the Annual Report to this prospectus supplement.
This prospectus supplement is not complete without the Prospectus. This prospectus supplement should be read in conjunction with the Prospectus, which is to be delivered with this prospectus supplement, and is qualified by reference thereto, except to the extent that the information in this prospectus supplement updates or supersedes the information contained in the Prospectus. Please keep this prospectus supplement with your Prospectus for future reference.
Our common stock is traded on the NASDAQ Capital Market under the symbol “ATNF”. On April 5, 2022, the last reported sale price for our common stock as reported on the NASDAQ Capital Market was $2.42 per share.
INVESTING IN OUR SECURITIES INVOLVES SUBSTANTIAL RISKS. SEE THE SECTION TITLED “RISK FACTORS” BEGINNING ON PAGE 7 OF THE PROSPECTUS TO READ ABOUT FACTORS YOU SHOULD CONSIDER BEFORE BUYING OUR SECURITIES.
NEITHER THE SECURITIES AND EXCHANGE COMMISSION NOR ANY STATE SECURITIES COMMISSION HAS APPROVED OR DISAPPROVED OF THESE SECURITIES OR PASSED UPON THE ADEQUACY OR ACCURACY OF THE PROSPECTUS OR THIS PROSPECTUS SUPPLEMENT. ANY REPRESENTATION TO THE CONTRARY IS A CRIMINAL OFFENSE.
The date of this prospectus supplement is April 6, 2022
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
☒ ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31, 2021
OR
☐ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from: __________ to __________
Commission File Number: 001-38105
180 LIFE SCIENCES CORP.
(Exact name of registrant as specified in its charter)
| Delaware | 90-1890354 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer |
|
3000 El Camino Real, Bldg. 4, Suite 200 |
94306 | |
| (Address of Principal Executive Offices) | (Zip Code) |
Registrant’s telephone number, including area code: (650) 507-0669
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered | ||
| Common Stock, par value $0.0001 per share | ATNF | The NASDAQ Stock Market LLC (NASDAQ Capital Market) | ||
| Warrants to purchase shares of Common Stock | ATNFW | The NASDAQ Stock Market LLC (NASDAQ Capital Market) |
Securities registered pursuant to Section 12(g) of the Act:
None.
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☐ No ☒
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☐ No ☒
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ☐ | Accelerated filer | ☐ |
| Non-accelerated filer | ☒ | Smaller reporting company | ☒ |
| Emerging growth | ☒ | ||
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
The aggregate market value of the voting and non-voting common stock held by non-affiliates of the registrant as of the last business day of the registrant’s most recently completed second fiscal quarter was approximately $271,883,250. For purposes of calculating the aggregate market value of shares held by non-affiliates, we have assumed that all outstanding shares are held by non-affiliates, except for shares held by each of our executive officers, directors and 5% or greater stockholders. In the case of 5% or greater stockholders, we have not deemed such stockholders to be affiliates unless there are facts and circumstances which would indicate that such stockholders exercise any control over our company, or unless they hold 10% or more of our outstanding common stock. These assumptions should not be deemed to constitute an admission that all executive officers, directors and 5% or greater stockholders are, in fact, affiliates of our company, or that there are not other persons who may be deemed to be affiliates of our company. Further information concerning shareholdings of our officers, directors and principal stockholders is incorporated by reference in Part III, Item 12 of this Annual Report on Form 10-K.
As of March 28, 2022, there were 34,087,244 shares of common stock issued and outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s definitive proxy statement relating to its 2022 annual meeting of shareholders (the “2022 Proxy Statement”) are incorporated by reference into Part III of this Annual Report on Form 10-K where indicated. The 2022 Proxy Statement will be filed with the U.S. Securities and Exchange Commission within 120 days after the end of the fiscal year to which this report relates.
| Auditor Firm Id: 688 | Auditor Name: Marcum, LLP | Auditor Location: San Francisco, CA |
TABLE OF CONTENTS
i
The following are abbreviations and definitions of certain terms used in this Report, which are commonly used in the pharmaceutical and biotechnology industry:
“ACA” means the Patient Protection and Affordable Care Act, often shortened to the Affordable Care Act, nicknamed Obamacare, which is a U.S. federal statute which provides numerous rights and protections that make health coverage fairer and easier to understand, along with subsidies (through “premium tax credits” and “cost-sharing reductions”) to make it more affordable. The law also expands the Medicaid program to cover more people with low incomes.
“Analgesics” are a class of medications designed specifically to relieve pain.
“ANDA” means an abbreviated new drug application which contains data which is submitted to the FDA for the review and potential approval of a generic drug product.
“Anti-TNF” is a pharmaceutical drug that suppresses the physiologic response to TNF.
“BLA” means the FDA’s Biologics License Application, which is the vehicle in the United States through which biologic sponsors formally propose that the FDA approve a new biologic for sale and marketing.
“Cannabinoids” mean compounds found in cannabis sativa L., and when used throughout this prospectus, refer to compounds found in the hemp plant which do not contain THC.
“CBD” or cannabidiol is an active ingredient in cannabis derived from the hemp plant. CBD is a non-psychoactive oxidative degradation product of THC.
“CBG” or cannabigerol is one of the compounds found in the cannabis plant.
“CCMO” means De Centrale Commissie Mensgebonden Onderzoek (CCMO), or the Central Committee on Research Involving Human Subjects, the organizational responsible for reviewing and regulating medical research involving human subjects in The Netherlands.
“CHMP” means the Committee for Medicinal Products for Human Use, formerly known as Committee for Proprietary Medicinal Products, which is the European Medicines Agency’s committee responsible for elaborating the agency’s opinions on all issues regarding medicinal products for human use.
“CMS” means the Centers for Medicare & Medicaid Services, which is a federal agency within the HHS that administers the Medicare program and works in partnership with state governments to administer Medicaid.
“Corticosteroids” are a class of drug that lowers inflammation in the body.
“CRO” means a contract research organization which is a company that provides support to the pharmaceutical, biotechnology, and medical device industries in the form of research services outsourced on a contract basis.
“CSA” means the Controlled Substances Act, the statute establishing federal U.S. drug policy under which the manufacture, importation, possession, use, and distribution of certain substances is regulated.
“CTA” means a Clinical Trial Application, which is a submission to the competent National Regulatory Authority(ies) for obtaining authorization to conduct a clinical trial in a specific country. It is an application with necessary information on investigational medicinal products. The purpose of a CTA is to provide all the important details about the clinical trial to the health authorities in order to obtain the product approval.
ii
“DEA” means the Drug Enforcement Administration, a United States federal law enforcement agency under the United States Department of Justice, tasked with combating drug trafficking and distribution within the United States.
“EMA” means the European Medicines Agency, an agency of the EU in charge of the evaluation and supervision of medicinal products.
“EU” means the European Union.
“FDC Act” means the Federal Food, Drug and Cosmetic Act, which is a set of U.S. laws passed by Congress in 1938 giving authority to the FDA to oversee the safety of food, drugs, medical devices, and cosmetics.
“FDA” means U.S. The Food and Drug Administration, which is a federal agency of the United States Department of Health and Human Services. The FDA is responsible for protecting the public health by ensuring the safety, efficacy, and security of human and veterinary drugs, biological products, and medical devices; and by ensuring the safety of U.S. food supply, cosmetics, and products that emit radiation.
“FS” means Frozen Shoulder, a condition characterized by stiffness and pain in an individual’s shoulder joint.
“GCP” means good clinical practice, which is an international quality standard, which governments can then transpose into regulations for clinical trials involving human subjects. GCP follows the International Council on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), and enforces tight guidelines on ethical aspects of clinical research.
“GLP” means good laboratory practice, which is a quality system concerned with the organization process and the conditions under which non-clinical health and environmental safety studies are planned, performed, monitored, recorded, archived and reported.
“GMP” means good manufacturing practice regulations promulgated by the FDA under the authority of the FDC Act. These regulations, which have the force of law, require that manufacturers, processors, and packagers of drugs, medical devices, some food, and blood take proactive steps to ensure that their products are safe, pure, and effective.
“HHS”, the U.S. Department of Health and Human Services also known as the Health Department, is a cabinet-level department of the U.S. federal government with the goal of protecting the health of all Americans and providing essential human services.
“HIPAA” means the Health Insurance Portability and Accountability Act of 1996, which has the goal of making it easier for people to keep health insurance, protect the confidentiality and security of healthcare information and help the healthcare industry control administrative costs.
“HMGB1” means High Mobility Group Box 1, a protein that, in humans, is encoded by the HMGB1 gene. Activated macrophages and monocytes secrete HMGB1 as a cytokine mediator of inflammation.
“IBD” means inflammatory bowel disease, an umbrella term used to describe disorders that involve chronic inflammation of the digestive tract.
“IND” means investigational new drug application. Before a clinical trial can be started, the research must be approved. An investigational new drug or IND application or request must be filed with the FDA when researchers want to study a drug in humans. The IND application must contain certain information, such as: results from studies so that the FDA can decide whether the treatment is safe for testing in people; how the drug is made, who makes it, what’s in it, how stable it is, and more; detailed outlines for the planned clinical studies, called study protocols, are reviewed to see if people might be exposed to needless risks; and details about the clinical trial team to see if they have the knowledge and skill to run clinical trials.
iii
“Individually identifiable health information” is defined by HIPPA to mean information that is a subset of health information, including demographic information collected from an individual, and: (1) is created or received by a health care provider, health plan, employer, or health care clearinghouse; and (2) relates to the past, present, or future physical or mental health or condition of an individual; the provision of health care to an individual; or the past, present, or future payment for the provision of health care to an individual; and (a) that identifies the individual; or (b) with respect to which there is reasonable basis to believe the information can be used to identify the individual.
“IRB” means an Institutional Review Board, which is group that has been formally designated to review and monitor biomedical research involving human subjects. In accordance with FDA regulations, an IRB has the authority to approve, require modifications in (to secure approval), or disapprove research. This group review serves an important role in the protection of the rights and welfare of human research subjects.
“Medicaid” is a federal and state health insurance program in the U.S. that helps with medical costs for some people with limited income and resources. Medicaid also offers benefits not normally covered by Medicare, including nursing home care and personal care services.
“Medicare” is a national health insurance program in the U.S. It primarily provides health insurance for Americans aged 65 and older, but also for some younger people with disability status as determined by the Social Security Administration, as well as people with end stage renal disease and amyotrophic lateral sclerosis (ALS or Lou Gehrig’s disease).
“MHRA” means The Medicines and Healthcare products Regulatory Agency, an executive agency of the Department of Health and Social Care in the United Kingdom which is responsible for ensuring that medicines and medical devices work and are acceptably safe.
“MRP” means a Mutual Recognition Procedure, a market authorization which is granted in one EU member state and is recognized in other EU member states.
“NDA” means the FDA’s New Drug Application, which is the vehicle in the United States through which drug sponsors formally propose that the FDA approve a new pharmaceutical for sale and marketing.
“NIHR” means The National Institute for Health Research is a United Kingdom government agency which funds research into health and care, and is the largest national clinical research funder in Europe.
“Orphan Drug Designation” means a pharmaceutical agent developed to treat medical conditions which, because they are so rare, would not be profitable to produce without government assistance.
“Phase 1” trials are typically where the drug is initially introduced into healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution and elimination. In the case of some drug candidates for severe or life-threatening diseases, such as cancer, especially when the drug candidate may be inherently too toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients.
“Phase 2” trials are generally when clinical trials are initiated in a limited patient population intended to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the drug candidate for specific targeted diseases and to determine dosage tolerance and optimal dosage. Phase 2 trials are sometimes further divided into: Phase 2a and Phase 2b trials — Phase 2a is focused specifically on dosing requirements. A small number of patients are administered the drug in different quantities to evaluate whether there is a dose-response relationship, which is an increase in response that correlates with increasing increments of dose. In addition, the optimal frequency of dose is also explored; and Phase 2b trials are designed specifically to rigorously test the efficacy of the drug in terms of how successful it is in treating, preventing or diagnosing a disease.
iv
“Phase 3” trials are when clinical trials are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk-benefit ratio of the drug candidate and provide an adequate basis for regulatory approval and product labeling.
“Phase 4” trials are studies required to be conducted as a condition of approval in order to gather additional information on the drug’s effect in various populations and any side effects associated with long-term use.
“Physiotherapy” is treatment to restore, maintain, and make the most of a patient’s mobility, function, and well-being.
“POCD” means post-operative cognitive dysfunction/delirium.
“RA” means rheumatoid arthritis.
“REMS” means a risk evaluation and mitigation strategy which is a drug safety program that the FDA can require for certain medications with serious safety concerns to help ensure the benefits of the medication outweigh its risks.
“SCA” means Synthetic Cannabidiol Analogs, which are synthetic pharmaceutical grade molecules close or distant analogs of non-psychoactive cannabinoids such as CBD for the treatment of inflammatory diseases and pain.
“Sponsor” means the applicant or drug sponsor, which is the person or entity who assumes responsibility for the marketing of a new drug, including responsibility for compliance with applicable provisions of the FSC Act and related regulations. Note that as used herein the term “Sponsor” may also refer to the Sponsor of our IPO, KBL IV Sponsor LLC, depending on the context in which such term is used.
“THC” means tetrahydrocannabinol, which is the principal psychoactive constituent of cannabis.
“TNF” means tumor necrosis factor, which is part of the body’s response to inflammation.
v
CAUTIONARY STATEMENT REGARDING FORWARD-LOOKING INFORMATION
This Annual Report on Form 10-K (this “Report”) contains forward-looking statements under federal securities laws, including within the meaning of the Private Securities Litigation Reform Act of 1995. In some cases, you can identify forward-looking statements by the following words: “anticipate,” “believe,” “continue,” “could,” “estimate,” “expect,” “intend,” “may,” “ongoing,” “plan,” “potential,” “predict,” “project,” “should,” or the negative of these terms or other comparable terminology, although not all forward-looking statements contain these words. Forward-looking statements are not a guarantee of future performance or results, and will not necessarily be accurate indications of the times at, or by, which such performance or results will be achieved. Forward-looking statements are based on information available at the time the statements are made and involve known and unknown risks, uncertainties and other factors that may cause our results, levels of activity, performance or achievements to be materially different from the information expressed or implied by the forward-looking statements in this Report, including those described under the heading “Risk Factors” contained in Item 1A of this Report. Forward-looking statements include, but are not limited to, statements about:
| ● | Expectations for the clinical and preclinical development, manufacturing, regulatory approval, and commercialization of our product candidates; | |
| ● | regulatory developments in the United States and foreign countries; |
| ● | our success in retaining or recruiting, or changes required in, our officers, key employees or directors; |
| ● | current negative operating cash flows and our potential ability to obtain additional financing to advance our business and the terms of any further financing, which may be highly dilutive and may include onerous terms; |
| ● | the continued impact of the COVID-19 pandemic on our business operations and our research and development initiatives; |
| ● | the accuracy of our estimates regarding expenses, future revenues and capital requirements; |
| ● | estimates of the sufficiency of our existing capital resources combined with future anticipated cash flows to finance our operating requirements; |
| ● | our ability to maintain our listing on Nasdaq; and |
| ● | other risks and uncertainties, including those listed under Part II, Item 1A. Risk Factors. |
Any forward-looking statements in this Report reflect our current views with respect to future events or to our future financial performance and involve known and unknown risks, uncertainties and other factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by these forward-looking statements. Given these uncertainties, you should not place undue reliance on these forward-looking statements. Except as required by law, we assume no obligation to update or revise these forward-looking statements for any reason, even if new information becomes available in the future.
vi
INTRODUCTION
General
The information included in this Annual Report on Form 10-K should be read in conjunction with the consolidated financial statements and related notes included at the end of this report.
Please see the “Glossary” above for a list of biotechnology industry abbreviations and definitions used throughout this Report.
Our logo and some of our trademarks and tradenames are used in this Report. This Report also includes trademarks, tradenames and service marks that are the property of others. Solely for convenience, trademarks, tradenames and service marks referred to in this Report may appear without the ®, ™ and SM symbols. References to our trademarks, tradenames and service marks are not intended to indicate in any way that we will not assert to the fullest extent under applicable law our rights or the rights of the applicable licensors if any, nor that respective owners to other intellectual property rights will not assert, to the fullest extent under applicable law, their rights thereto. We do not intend the use or display of other companies’ trademarks and trade names to imply a relationship with, or endorsement or sponsorship of us by, any other companies.
The market data and certain other statistical information used throughout this Report are based on independent industry publications, reports by market research firms or other independent sources that we believe to be reliable sources. Industry publications and third-party research, surveys and studies generally indicate that their information has been obtained from sources believed to be reliable, although they do not guarantee the accuracy or completeness of such information. We are responsible for all of the disclosures contained in this Report, and we believe these industry publications and third-party research, surveys and studies are reliable. While we are not aware of any misstatements regarding any third-party information presented in this Report, their estimates, in particular, as they relate to projections, involve numerous assumptions, are subject to risks and uncertainties, and are subject to change based on various factors, including those discussed under the section entitled “Risk Factors” of this Report. These and other factors could cause our future performance to differ materially from our assumptions and estimates. Some market and other data included herein, as well as the data of competitors as they relate to 180 Life Sciences Corp., is also based on our good faith estimates.
Our fiscal year ends on December 31. Interim results are presented on a quarterly basis for the quarters ended March 31, June 30, and September 30, the first quarter, second quarter and third quarter, respectively, with the quarter ended December 31st being referenced herein as our fourth quarter. Fiscal 2021 means the year ended December 31, 2021, whereas fiscal 2020 means the year ended December 31, 2020.
Definition
Unless the context requires otherwise, references to the “Company,” “we,” “us,” “our,” “180 Life”, “180LS” and “180 Life Sciences Corp.” refer specifically to 180 Life Sciences Corp. and its consolidated subsidiaries. References to “KBL” refer to the Company prior to the November 6, 2020 Business Combination (discussed and defined below).
In addition, unless the context otherwise requires and for the purposes of this Report only:
| ● | “CAD” refers to Canadian dollars; | |
| ● | “Exchange Act” refers to the Securities Exchange Act of 1934, as amended; | |
| ● | “£” or “GBP” refers to British pounds sterling; | |
| ● | “SEC” or the “Commission” refers to the United States Securities and Exchange Commission; and | |
| ● | “Securities Act” refers to the Securities Act of 1933, as amended. |
1
Where You Can Find Other Information
We file annual, quarterly, and current reports, proxy statements and other information with the SEC. Our SEC filings are available to the public over the Internet at the SEC’s website at www.sec.gov and are available for download, free of charge, soon after such reports are filed with or furnished to the SEC, on the “Investors”—“SEC Filings”—“All SEC Filings” page of our website at www.180lifesciences.com. Copies of documents filed by us with the SEC are also available from us without charge, upon oral or written request to our Secretary, who can be contacted at the address and telephone number set forth on the cover page of this Report. Our website address is www.180lifesciences.com/. The information on, or that may be accessed through, our website is not incorporated by reference into this Report and should not be considered a part of this Report.
Company Overview
We are a clinical stage biotechnology company headquartered in Palo Alto, California, focused on the development of therapeutics for unmet medical needs in chronic pain, inflammation and fibrosis by employing innovative research, and, where appropriate, combination therapy. Our Company was founded by several world-leading scientists in the biotechnology and pharmaceutical sectors. Our world-renowned scientists Prof. Sir Marc Feldmann, Prof. Lawrence Steinman, Prof. Raphael Mechoulam, Dr. Jonathan Rothbard, and Prof. Jagdeep Nanchahal have significant experience and significant previous success in drug discovery. The scientists are from the University of Oxford (“Oxford”), Stanford University and Hebrew University of Jerusalem (the “Hebrew University”), and the management team has extensive experience in financing and growing early-stage healthcare companies.
We have three different product development platforms that are focused on different diseases or medical conditions, and that target different factors, molecules or proteins, as follows:
| ● | Anti-TNF platform: focusing on fibrosis and anti-tumor necrosis factor (“anti-TNF”); |
| ● | SCAs platform: focusing on drugs which are synthetic cannabidiol (“CBD”) or cannabigerol (“CBG”) analogues (“SCAs”); and |
| ● | α7nAChR platform: focusing on alpha 7 nicotinic acetylcholine receptor (“α7nAChR”). |
Our lead product candidate recently completed a successful Phase 2b clinical trial in the United Kingdom (“U.K.”) and the Netherlands for early-stage Dupuytren’s Contracture, a condition that affects the development of fibrous connective tissue in the palm of the hand. On December 1, 2021 the Company announced top line data from the trial, which indicates that the primary end point of nodule hardness and the secondary end point of nodule size on ultrasound scan were met with statistically significant differences. There were no related severe adverse events. The full results have been submitted for publication in a peer-reviewed journal and will be disclosed upon publication.
Currently, we are conducting clinical trials only for certain indications under the anti-TNF platform. Of our three product development platforms, only one, the SCAs platform, involves products that are related to CBD (and not to cannabis or THC), and no clinical trials for any indications or products under the SCAs platform are currently being conducted in the United States or abroad. We are currently undertaking preclinical research and development activities for the SCA and the α7nAChR platforms.
2
Business Strategy
Our goal is to capitalize on our research in chronic pain, inflammation and fibrosis by pursuing the following strategies:
| ● | advance our clinical-stage product candidate for early-stage Dupuytren’s Contracture from its current late-stage development to seek and obtain approval in the U.K., European Union (“EU”) and the United States (“U.S.”) for such product candidate, potentially commercialize the product candidate in the U.K., EU and U.S. and identify the optimal commercial pathway in other markets around the world; |
| ● | move our pre-clinical product candidates into clinical trials, seek and obtain approval in the U.K., EU and U.S. for such future product candidates, and potentially commercialize such future product candidates in the U.S., U.K. and EU; |
| ● | leverage our proprietary product development platforms to discover, develop and commercialize novel first-in-class products for the treatment of chronic pain, inflammation and fibrosis; and |
| ● | strengthen our position in research in chronic pain, inflammation and fibrosis. |
Overview of Product Development Platforms
The following chart summarizes the products and indications, including those currently in clinical trial, for our three product development platforms.
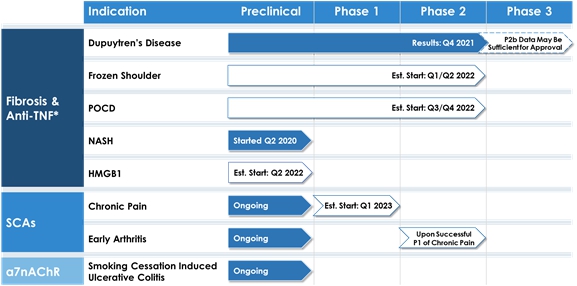
“*Regulatory approvals obtained from the MHRA and CCMO and the relevant accredited ethics committees to perform clinical trials in the U.K. and The Netherlands. No meetings have been held with, and no applications or requests for approval have been submitted to the FDA for any products at this time.”
On December 1, 2021, the Company announced positive top line data for the Phase 2b clinical trial of Dupuytren’s Contracture.
The product development platforms are each described in more detail below.
3
Fibrosis & Anti-TNF Platform
Our anti-TNF platform began at our wholly-owned subsidiary, 180 LP. This platform is focused on studying the molecular mechanism of inflammatory diseases and fibrosis and on the discovery of TNF as a mediator of fibrosis, as well as other immune-driven diseases. This research was first undertaken in the 1980s by our Co-Chairman, Prof. Sir Marc Feldmann, based on analysis of tissue from patients with rheumatoid arthritis (“RA”). We are applying this same approach to the analysis of human disease tissue from patients with active fibrosis, research led by Prof. Jagdeep Nanchahal in Oxford (who is also the Chairman of our Clinical Advisory Board), which has led to the identification of new therapeutic targets and approaches that we are developing. Profs. Nanchahal and Feldmann, in collaboration with other scientists, are leveraging their experience and expertise in developing anti-inflammatories to search for new applications for anti-TNF therapeutics. We are seeking to demonstrate that anti-TNF drugs, such as adalimumab, have a positive effect on new indications such as Dupuytren’s Contracture, frozen shoulder and post-operative cognitive dysfunction/delirium (“POCD”).
Our first product candidate in clinical development is for the potential treatment of early-stage fibrosis of the hand, Dupuytren’s Contracture, for which there is currently no approved treatment in the U.K. or EU. Collagenase from Clostridium histolyticum has been approved in the USA for late-stage Dupuytren’s Contracture. The proposed treatment will be administered by a local injection of adalimumab, an anti-TNF antibody, into early-stage disease tissue. The results for the Phase 2a clinical trial for Dupuytren’s Contracture, supported by the Wellcome Trust, U.K. Department of Health and the Company, were published in July 2018. The study demonstrated positive tissue response indicative of anti-fibrotic mechanisms, as well as guiding dosing for follow up trials. Having defined the most efficacious dose and preparation and based on these positive proof of concept data, the Company, together with the Wellcome Trust and the U.K. Department of Health, initiated a Phase 2b trial in patients with early-stage Dupuytren’s Contracture. The initial plan was to randomize 138 patients in a ratio of 1:1 to receive four injections of adalimumab or placebo at three-month intervals, and followed for a total of 18 months from baseline. With additional funding from the Wellcome Trust, the Phase 2b trial completed recruitment of 174 patients in April 2019, having commenced dosing in February 2017. The final patient was enrolled in April 2019. The Phase 2b clinical trial for early-stage Dupuytren’s Contracture has been completed. On December 1, 2021, the Company announced top line data from the trial, which indicates that the primary end point of nodule hardness and the secondary end point of nodule size on ultrasound scan were met with statistically significant differences. There were no related severe adverse events. The full results have been submitted for publication in a peer-reviewed journal and will be disclosed upon publication. Through this fibrosis and anti-TNF product development platform, we are also performing research for the development of potential treatments of frozen shoulder, liver and lung fibrosis and POCD.
The following chart summarizes the timing of current and future clinical trials, based on current proposals, under the anti-TNF platform.
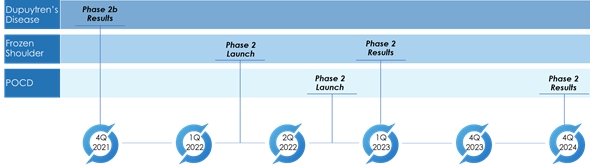
We have obtained regulatory approvals from the U.K. Medicines and Healthcare Products Regulatory Agency (MHRA) and the Dutch Centrale Commissie Mensgebonden Onderzoek (CCMO), as well as from the relevant accredited ethics committees, in order to perform clinical trials in the U.K. and The Netherlands solely for indications under the anti-TNF platform. We have not held any meetings with, and no applications or requests for approval have been submitted to, the U.S. Food and Drug Administration (“FDA”) for any indications or products under the anti-TNF platform at this time.
4
HMGB1 Program
Our HMGB1 program was recently formed with the in-licensing of the technology from the University of Oxford on November 2, 2021. Our HMGB1 program falls under the Fibrosis and Anti-TNF Platform. We have identified HMGB1 as a therapeutic target that acts on multiple endogenous adult stem cells to accelerate the physiological regenerative response to current or future injuries. These findings have broad relevance to the fields of stem cell biology and regenerative medicine and suggest a therapeutic approach to promote tissue repair such as in NASH liver regeneration.
The technology was developed by Prof. Jagdeep Nanchahal’s laboratory at the University Oxford prior to licensing. Due to the early stage of development of HMGB1, we are still in the process of assessing milestones and development timelines. The licensing of HMGB1 included the lead development candidate for liver fibrosis.
No regulatory approvals have been sought or obtained from appropriate authorities at this time for any products or indications under the HMGB1 platform.
SCAs Platform
Our SCAs platform began at our wholly-owned subsidiary, CBR Pharma with the collaborative work of its founders Prof. Mechoulam and Prof. Feldmann. This platform focuses on the development of synthetic pharmaceutical grade molecules close or distant analogs of non-psychoactive cannabinoids such as CBD for the treatment of inflammatory diseases and pain. These development efforts are a result of a 20-year collaboration between Prof. Feldmann, who discovered and commercialized anti-TNF therapy for treatment of RA and subsequently a number of inflammatory diseases, which is currently the best-selling drug class in the world, and Prof. Mechoulam, a world leading expert in cannabis chemistry who successfully identified THC, CBD and, subsequently, the endocannabinoids. We are working with a research team based at the Kennedy Institute at Oxford, consisting of Prof. Feldmann, Prof. Richard Williams and others, and a research team based at Hebrew University, consisting of Prof. Raphael Mechoulam, Prof. Avi Domb, Prof. Amnon Hoffman and others, to generate new drugs, test them, and optimize their uptake and delivery to disease targets. The aim is to develop novel, orally active analgesic and anti-inflammatory medications based on synthetic compounds to target chronic diseases. We term these synthetic compounds generically as “synthetic CBD analogs” (“SCAs”). Our primary development targets are arthritis and chronic and recurrent pain, while our secondary development targets are diabetes/diabetic neuropathy, fibromyalgia, multiple sclerosis, obesity and fatty liver disease.
The following chart summarizes the timing of current and future clinical trials, based on current proposals, under the SCAs platform.

No regulatory approvals have been sought or obtained from appropriate authorities at this time for any products or indications under the SCAs platform.
5
α7nAChR Platform
Our α7nAChR platform began at our wholly-owned subsidiary, Katexco, where its founders identified α7nAChR as a key receptor for the amyloid proteins associated with diseases like Alzheimer’s and Parkinson’s Disease. α7nAChR is expressed on the surface of both neuronal cells in the brain and on important cells of the immune system. The research conducted by Dr. Jonathan Rothbard and Prof. Steinman has shown that small molecules available as drugs taken by mouth can engage this receptor and potently reduce inflammatory diseases. Dr. Rothbard and Prof. Steinman have also shown that α7nAChR is critical in reducing disease animal models of multiple sclerosis and RA, as well as heart attack and stroke. Our α7nAChR product development platform is currently focused on developing α7nAChR agonists for the treatment of inflammatory diseases, initially ulcerative colitis induced after cessation of smoking.
No regulatory approvals have been sought or obtained from appropriate authorities at this time for any products or indications under the α7nAChR platform.
Product Candidates
We are attempting to build a broad and diverse pipeline of product candidates in chronic pain, inflammation and fibrosis. Our product candidates are and will be selected for development based on: potential to address unmet medical needs; development feasibility as determined by our preclinical research and development efforts; potential to rapidly achieve proof-of-concept based on easy-to-measure validated regulatory endpoints; and significant commercial potential.
Anti-TNF Platform Dupuytren’s Contracture
Overview
Dupuytren’s Contracture, also referred to as hand fibrosis, is a progressive, incurable disease characterized by the development of fibrous cords in the palm of the hand, commonly affecting the ring and/or small finger and often multiple joints, leading to contracture and the inability to straighten the affected fingers. Symptoms, when presented to a physician, range from the appearance of nodules in the palm, which can be painless or painful and often disconcerting to the patient, to the loss of the use of the contracted finger. There are currently no approved treatment options for those patients who present with symptomatic, early-stage disease.
Surgery remains the standard treatment for patients with Dupuytren’s Contractures but is associated with extended recovery periods and risks of recurrence.
We are developing therapies by repurposing of the anti-TNF therapeutic adalimumab, previously approved and used under the brand name Humira for several autoimmune conditions, for the treatment of early-stage Dupuytren’s Contracture. Research at Oxford University has indicated an anti-TNF mechanism can slow or prevent the proliferation of myoblast cells that lead to the formation and growth of the fibrous nodules/cords in the palm and possible finger contracture. We have advanced the development program through Phase 2b clinical trials to evaluate the impact of multiple, intralesional injections on disease progression and functional improvement.
Dupuytren’s patients who have advanced disease are primarily treated by orthopedic or plastic surgeons, who rely on invasive interventions when the contracture impacts hand function. Current treatment options include open surgeries (fasciotomies or fasciectomies) and the less invasive procedures of needle aponeurotomy (NA) or collagenase injections. The less invasive procedures are designed to disrupt the integrity of a contracted cord so the fingers can be straightened. Unfortunately, these options are associated with a high rate of recurrence. Dissatisfaction within the medical and Dupuytren patient community with outcomes for later-stage disease and the lack of options to intervene at an early/pre-contracture stage indicate there is an unmet medical need for early-stage intervention.
According to the Dupuytren’s Foundation, Dupuytren’s Contracture prevalence is estimated to be up to 7% of the U.S. population. Based on the Foundation’s estimates, approximately three million patients have contractures that should be treated but only 10% to 20% of those patients are treated. Reasons for the lack of treatment may include the type of available interventions, poor long-term outcomes, and reimbursement hurdles.
6
In primary interviews in late 2021 with 8 orthopedic/plastic surgeons, conducted by Red Sky Partners (an independent third-party consulting firm) on our behalf and designed to better understand the unmet need for patients with Dupuytren’s Contracture, revealed a strong desire among hand surgeons and patients to treat this condition early, before the development of late stage contractures, in a non-invasive manner that will limit further progression, preserve function and prevent or delay invasive surgery. Surgeons’ reactions to the rationale for the use of adalimumab to address this unmet need were overall positive and the mechanistic concept of an anti-TNF compound was considered compelling. In the view of the majority of surveyed hand surgeons, the non-invasive, safe product profile would potentially position adalimumab as an important therapeutic option for a much wider range of patients than are typically treated today. Assuming clinical efficacy and safety are supported with published data, we believe that adalimumab would become an attractive alternative to surgery, needle aponeurotomy or collagenase. Further, we believe it has potential use in many early-stage patients who are not treated today.
Based on both primary (feedback from these physician interviews) and secondary research, Red Sky Partners concluded that an initial label focused on patients with a clear contracture where adalimumab would soften nodules and limit progression would be highly differentiated from current therapies and could generate revenues in the range of $300 million to $350 million annually in the U.S. More significantly, the opportunity to offer a safe, non-invasive therapeutic leading to improved function could dramatically expand the treatable population as more patients seek treatment and more physicians are motivated to offer their patients an alternative to waiting to see if their disease progresses, which they cannot do today. This product positioning could generate a revenue opportunity two to three times the initial market opportunity.
Phase 2 Clinical Trials
Our wholly-owned subsidiary, 180 LP, contributed to the funding of a Phase 2a clinical trial for Dupuytren’s Contracture along with the Wellcome Trust and the U.K. Department of Health, which using an experimental medicine clinical trial design demonstrated positive tissue response, as well as guiding dosing and tolerability for follow-up trials. The data was published in June 2018.
For the Phase 2a trial, we recruited 28 patients, eight assigned to the 15 milligrams (mg), 12 to the 35 mg and eight to the 40 mg adalimumab cohorts. There was no change in mRNA levels for ACTA2, COL1A1, COL3A1 and CDH11. Levels of α-SMA protein expression in patients treated with 40 mg adalimumab (1.09 ± 0.09 ng per μg of total protein) were significantly lower (p = 0.006) compared to placebo treated patients (1.51 ± 0.09 ng/μg). The levels of procollagen type I protein expression were also significantly lower (p = 0.019) in the sub group treated with 40 mg adalimumab (474 ± 84 pg/μg total protein) compared with placebo (817 ± 78 pg/μg). There were two serious adverse events, both considered unrelated to the study drug. In this dose-ranging study, injection of 40 mg of adalimumab in 0.4 ml resulted in down regulation of the myofibroblast phenotype as evidenced by reduction in expression of α-SMA and type I procollagen proteins at 2 weeks.
Having defined the most efficacious dose and preparation and based on these positive proof-of-concept data, the Company, together with the Wellcome Trust and the U.K. Department of Health, initiated a Phase 2b trial in patients with early-stage Dupuytren’s Contracture. The initial plan was to randomize 138 patients in a ratio of 1:1 to receive four injections of adalimumab or placebo at three-month intervals and followed for a total of 18 months from baseline. The Phase 2b trial, which was funded by grants from the Wellcome Trust and the U.K. Department of Health, with a contribution from 180 LP to purchase the drug, completed recruitment of 174 patients in April 2019 and commenced dosing in February 2017 in the U.K. and Groningen, The Netherlands.
The Phase 2b clinical trial for early-stage Dupuytren’s Contracture has been completed. On December 1, 2021, the Company announced top line data from the trial, which indicates that the primary end point of nodule hardness and the secondary end point of nodule size on ultrasound scan were met with statistically significant differences. There were no related severe adverse events. The full results have been submitted for publication in a peer-reviewed journal and will be disclosed upon publication.
7
Other Product Candidates or Indications
In addition to the potential treatment, we are developing for Dupuytren’s Contracture described above, we are seeking to repurpose anti-TNF for use as a treatment for other fibrotic conditions such as frozen shoulder. Prof. Feldmann’s previous work in the 1980s demonstrated that anti-TNF is an effective anti-inflammatory with many possible uses, and it was subsequently approved for various forms of inflammatory arthritis and inflammatory bowel disease (IBD), as well as other indications. This has since created what is currently the best-selling drug class in the world, anti-TNF therapeutics, which, according to Research and Markets, was valued at over $40 billion in 2019. By using a well-known and extensively used therapeutic, adalimumab, the research and development process may be truncated because of existing product information relating to safety, as the drug has been widely used over the past 20 years in millions of patients.
Frozen Shoulder
Frozen shoulder, also referred to as adhesive capsulitis, is an extremely painful and debilitating condition that affects an individual’s everyday activities, including sleep. Frozen shoulder is estimated to affect approximately 9% of the population in Western countries between the ages of 25 and 64 according to an article published in Arthritis & Rheumatology in 2004. In addition, approximately 20% of people suffering from a frozen shoulder will develop the same problem in their other shoulder. According to an article published in Shoulder & Elbow in 2010, it is estimated that up to 30% of patients with diabetes develop frozen shoulder, and the symptoms tend to be more persistent and recalcitrant in this group.
During the pain predominant inflammatory phase, patients are typically treated with analgesics, physiotherapy and corticosteroid injections. Patients with persistent stiffness may be referred to secondary care for capsular release by manipulation under anesthesia, hydrodilatation or surgical arthroscopy. To our knowledge, there is currently no approved targeted therapy, and in conjunction with the National Institute for Health Research (U.K.), we are investigating the feasibility of recruiting patients during the early pain-predominant inflammatory phase of the disease and delivery of a local injection of anti-TNF. The set-up stage for this Phase 2 clinical trial for the local injection of anti-TNF for frozen shoulder started in June 2021. A £250,000 grant has been awarded from NIHR to the University of Oxford to support execution and clinical trial sites are being identified. The Company will provide additional funding to support this trial. All the requisite regulatory approvals are in place and the investigators expect to administer the first patient for the frozen shoulder Phase 2 clinical trial in the first or second quarter of 2022.
Human Liver Fibrosis
Fibrosis of the liver is characterized by long-term damage to the organ caused by the replacement of normal liver tissue with scar tissue. The condition is most commonly caused by non-alcoholic fatty liver disease (“NAFLD”), which encompasses non-alcoholic fatty liver (“NFL”) and non-alcoholic steatohepatitis (“NASH”). NAFLD affects approximately 30% of the U.S. population, according to an article published in Nature Reviews Gastroenterology & Hepatology in 2016. Approximately 2% of patients with NFL and approximately 15% to 20% of patients with NASH progress to cirrhosis, fibrosis of the liver with major health issues.
To our knowledge, there is no current approved treatment for individuals with NASH. We therefore believe that there is a large potential market for the creation of an effective preventative treatment. According to Allied Market Research, the market for treating liver fibrosis was approximately $13 billion in 2018, and is projected to rise to approximately $20 billion in 2022, rising at a CAGR of over 11% per year. We initiated preclinical studies for NASH based on human liver samples during the second quarter of 2020.
Post-operative Cognitive Decline (POCD)
POCD is a common neuropsychiatric syndrome, defined as disturbance of attention, awareness and cognition, which develops over a short period of time and tends to fluctuate during the course of the day. Patients with hip fracture are at particularly high risk of developing POCD. The U.K.’s national audit data for 2018 showed that 25% of all patients with hip fracture suffered from delirium. POCD is associated with poor functional outcomes, reduced quality of life and longer hospital stays. People with hip fracture who developed delirium are twice as likely to die as inpatients, and nearly four times more likely to need placement in a nursing home. POCD has also been closely associated with long-term cognitive impairment.
8
Hip fractures are one of the main challenges facing elderly patients and healthcare systems. According to an article published in The Lancet Public Health in 2017, hip fractures are associated with an average loss of 2.7% of the healthy life expectancy in the middle-aged and older population in the U.S. and Europe. People suffering hip fracture have a mean age of 83 years, are frail, and two-thirds are women. They suffer a 30-day mortality of 7%, and experience a persistent reduction in their health-related quality-of-life similar to that of a diagnosis of Parkinson’s disease or multiple sclerosis. According to various studies, POCD is developed in 13-40% of patients following cardiac surgery. With 500,000 open heart surgeries and 450,000 hip surgeries in the USA each year, in advanced age patients, a beneficial therapy to treat POCD would be a significant benefit to these patients. We plan to initiate a Phase 2 study using anti-TNF for POCD during the second quarter of 2022. An issued patent to protect this potential use has been licensed from The Kennedy Trust for Rheumatology Research.
SCAs Platform
Overview
Cannabinoids are a class of compounds derived from cannabis plants. The two major cannabinoids contained in cannabis are CBD and THC. Although one cannabinoid, THC, is known to cause psychoactive effects associated with the use of herbal cannabis, no other cannabinoid is known to share these properties. In recent decades, there have been major scientific advances that have led to the discovery of new plant-derived cannabinoids and the endocannabinoid system. There are at least two types of cannabinoid receptors in the human endocannabinoid system, cannabinoid receptor 1 (“CB1”) and cannabinoid receptor 2 (“CB2”). CB1 receptors are considered to be among the most widely expressed G protein-coupled receptors in the brain and are particularly abundant in areas of the brain concerned with movement and postural control, pain and sensory perception, memory, cognition, emotion, and autonomic and endocrine function. CB1 receptors are also found in peripheral tissues including peripheral nerves and non-neuronal tissues such as muscle, liver tissues and fat. CB2 receptors are expressed primarily in tissues in the immune system and are believed to mediate the immunological effects of cannabinoids. CBD does not interact with CB1 receptors and is only a weak agonist of CB2 receptors. CBD interacts with other important neurotransmitter and neuromodulatory systems in the human body, including transient receptor potential channels, adenosine uptake and serotonin receptors. The far-reaching and diverse pharmacology of the numerous cannabinoids provides significant potential for development of cannabinoid therapeutics across many indications and disease areas, but also adds to the complexity of the research.
For the SCA program, we have agreements in place with Hebrew University and Oxford, pursuant to which we intend to conduct research to develop and characterize novel SCAs for the treatment of certain target indications, and to perform early-phase clinical trials. Through the Research Agreements with Hebrew University and Oxford, we established research facilities at the Hebrew University and Oxford, in which the development and testing of new cannabinoids designed and synthesized at the Hebrew University will be facilitated. The labs at the Hebrew University, led by Prof. Mechoulam, will synthesize the chemical compounds and perform preliminary efficacy and safety studies.
Once these initial studies are completed at the Hebrew University, the chemical compounds are sent to Prof. Richard Williams at Oxford, where further evaluation is carried out to identify candidates which have the best potential for clinical efficacy and commercial development. Subsequently, we will support the clinical development of the lead compound(s), culminating in Phase 2 clinical trials to establish clinical utility in chronic pain and inflammatory indications.
The initial focus of the research will be on the development of safe and well-tolerated compounds with analgesic and immunomodulatory activity and with the capacity to synergize with current therapies, which primarily target downstream inflammatory processes. After conducting initial research and development, we will select the most promising of the chemical compounds to move into Phase 1 and 2 clinical trials, which we expect to commence by the third quarter of 2022.
9
Product Candidates or Indications
We believe that there are unmet needs for orally available, relatively safe anti-inflammatory drugs, especially those with analgesic properties. We believe that SCAs have the potential to fulfill these needs and we have started to develop novel, orally available and patentable drug candidates to treat certain diseases or conditions such as arthritis, multiple sclerosis, diabetes, psoriasis, obesity and fatty liver, and various painful conditions. Our work on SCAs is currently in the preclinical development stage.
Because medical cannabis is a complex mixture of compounds from plants, providing a consistent level of the active compound of interest or controlling the level of the other natural compounds is difficult. Accordingly, we are working on orally available SCAs, not derived from plants, to address the deleterious issues of medical cannabis described above. If successful, these SCAs could become approved drug products that offer a robustly consistent and safe dosage that allows patient intake to be carefully controlled.
We believe that the development and clinical study of SCAs will reveal that SCAs have several key advantages over medical cannabis, including:
| ● | use of a pure compound (>99.5%) rather than a mixture of compounds; |
| ● | ability to test and control dosing, which in turn controls efficacy and side effect levels; |
| ● | creation of a reproducible product; and |
| ● | ability to engineer novel synthetic analogs to control binding preferences to select receptors, control agonist or antagonist effects of receptor binding (pharmacokinetics and dynamics), modify half-life of the drug in the body, and create pro-drug forms that are only activated in specified tissues, thereby potentially reducing off target side effects. |
In addition to the above advantages, testing SCAs in scientific, double-blind clinical trials would help to allay physicians’ concerns regarding the therapeutic use of marijuana-based compounds. This change could increase the number of patients that have access to these drug therapies. If clinical trials are successful, there are a number of potential markets and indications for SCAs which we could target, which include individuals suffering from chronic and recurrent pain, diabetes, osteoarthritis, obesity and fatty liver disease.
α7nAChR Platform
Overview
Two of our lead scientists, Prof. Steinman and Dr. Rothbard, previously identified a key receptor for the amyloid proteins associated with diseases like Alzheimer’s and Parkinson’s disease, called α7nAChR. The α7nAChR is expressed on the surface of both neuronal cells in the brain and on cells of the immune system. The research conducted by Dr. Rothbard and Prof. Steinman has shown that small molecules available as drugs taken by mouth can engage this receptor and potently reduce inflammatory diseases. Dr. Rothbard and Prof. Steinman have shown that this receptor is critical in reducing disease in animal models of multiple sclerosis and RA, as well as heart attack and stroke.
Our efforts to understand the role of the high concentration of small heat shock proteins (“sHsp”) found in the lesions in the brains of patients with multiple sclerosis led us to realize that the protein was (i) immune suppressive and (ii) therapeutic in animal 2 models of multiple sclerosis, cardiac and retinal ischemia, and stroke. A significant realization was that amyloid fibrils composed of proteins or small peptides exhibited biological responses equivalent to the sHsps. The fibrils and the sHsps specifically bound and activated macrophages (“MΦ”) and regulatory B cells. Crosslinking and precipitation experiments demonstrated that both species bound nAChR and signaled through Jak2/Stat3. We realized that nicotine treatment of experimental autoimmune encephalomyelitis (“EAE”) induces an identical pattern of immune suppression as our treatments and exhibits pre-clinical efficacy that is comparable with many of the drugs that are approved for multiple sclerosis (MS) when they were tested in EAE models. Collectively, these observations have informed our strategy to develop an orally available, small molecule agonist of α7nAChR for inflammation and autoimmune diseases.
10
The α7 subunit of α7nAChR is an integral part of an endogenous immune suppressive pathway, in which activation of the vagus nerve stimulates acetylcholine secretion, which in turn binds α7nAChR on MΦs and regulatory B lymphocytes. Activation of the MΦs initiates an immunosuppressive cascade of events that lead to reduction of pro-inflammatory cytokines, suppression of B and T cell activation and control of inflammation.
In autoimmune diseases like RA, where there is intense inflammation destroying joints, and in multiple sclerosis, where the brain is under attack with damage to vital neurologic circuits, the body’s immune system turns against its own tissues. Other diseases ranging from atherosclerosis to gout, also reveal manifestations of an unwanted autoimmune attack.
Activation of the α7nAChR results in a signaling cascade involving Jak2 and Stat3 leading to the conversion of the macrophages to an immune suppressive phenotype and the production of IL-10. IL-10 is known to reduce inflammatory cytokines, most prominently TNF, IL-1, and IL-6. Consequently, α7nAChR agonists should complement anti-TNF therapy, which opens up the possibility of developing a new class of orally available medicines which are anti-inflammatory but much safer than existing medications such as NSAIDS, Cox2 inhibitors, methotrexate, and Janus kinase (JAK) inhibitors. This is because α7nAChR agonists are activating an endogenous regulatory pathway, rather than blocking important pathways needed for diverse processes. The market opportunity arises from the complex and expensive effort by several large and small biotechnology companies in the development of a spectrum of orally available partial agonists specific for α7nAChR. The compounds underwent extensive preclinical assessment and were used in 18 studies comprising 2,670 subjects.
The drugs universally were shown to be safe, but ineffective in trials for neurologic and psychiatric diseases, namely Alzheimer’s disease and schizophrenia. In randomized, placebo-controlled clinical trials for cognitive impairment in Alzheimer’s disease and schizophrenia, the compounds failed to meet their primary endpoint.
We plan to use these previous studies as a foundation to potentially develop a patentable α7nAChR analog within this family to use as an immune suppressive to treat a range of inflammatory and autoimmune indications including RA, inflammatory bowel disease (IBD), relapsing and progressive forms of multiple sclerosis, atherosclerosis, gout and osteoarthritis. Our scientists have found that the α7 receptor on macrophages and regulatory B lymphocytes are different from the target of the drugs developed so far.
Product Candidates or Indications
We intend to identify, characterize, synthesize, and patent an orally available small molecular weight agonist of α7nAChR by screening non-patented analogs of large numbers of known agonists defined by pharmaceutical companies. We intend to outsource this work to Evotec GMBH, an integrated early discovery organization, and one which we have worked with in the past, specializing in ion channels and transporters, offering clients specialized technologies and scientific expertise to move from target to lead compounds.
Following a safety and efficacy assessment program, we intend to select candidates for pre-clinical development as a prelude to the potential initiation of clinical studies, which could potentially be followed by an Investigational New Drug Application (“IND”) to the FDA. Our first intended target indication for its α7nAChR development platform is smoking cessation induced ulcerative colitis.
11
Outsourcing and Manufacturing
We are currently outsourcing our clinical trials, which are conducted at Oxford University, Edinburgh, U.K. and Groningen, The Netherlands and only involve certain indications under the anti-TNF platform. We expect to continue to outsource our clinical trials and conduct them at (1) in the case of the anti-TNF platform, Oxford University and Groningen, The Netherlands, (2) in the case of the SCAs platform, Hebrew University and Oxford University, and (3) in the case of the α7nAChR platform, to be determined.
We also expect to outsource all of our manufacturing activities, including those activities at the research or clinical stage, with SCAs to be produced at Hebrew University and α7nAChR to be produced by Evotec GMBH and the anti-TNF platform utilizing off-the-shelf adalimumab. In addition, we expect our products to be good manufacturing practice (GMP) grade and produced by accredited contract research organizations (CROs).
Material Agreements
We have entered into material research and licensing agreements (the “Research Agreements”) with various universities and parties in order to conduct research to develop potential product candidates. We have also entered into other material consulting and advisory services agreements with various scientists (the “Consulting Agreements”) to assist with such research.
Overview of Research Agreements
The Research Agreements include agreements with the Hebrew University and Oxford. For the anti-TNF platform, the Research Agreements with Oxford allow the Company to contribute financially to sponsor the research being conducted for the anti-TNF platform. In return, the Company will receive an exclusive option to license any intellectual property arising from the Research Agreements. There are also license agreements in place whereby we have exclusively licensed certain intellectual property from Oxford.
For the SCA program, we have agreements in place with Hebrew University and Oxford, pursuant to which we intend to conduct research to develop and characterize novel SCAs for the treatment of certain target indications, and to perform early-phase clinical trials. Through the Research Agreements with Hebrew University and Oxford, we established research facilities at the Hebrew University and Oxford, in which the development and testing of new cannabinoids designed and synthesized at the Hebrew University will be facilitated.
The Research Agreements are each described below.
Research Agreements with the Hebrew University
On May 13, 2018, our wholly-owned subsidiary CBR Pharma entered into a research and license agreement (the “2018 Hebrew Agreement”) with Yissum Research Development Company of the Hebrew University of Jerusalem, Ltd. (“Yissum”), pursuant to which Yissum granted CBR Pharma a worldwide exclusive license (the “2018 Hebrew License”) to develop and commercialize certain patents (the “2018 Hebrew Licensed Patents”), know-how and research results (collectively, the “2018 Hebrew Licensed Technology”), in order to develop, manufacture, market, distribute or sell products, all within the use of the 2018 Hebrew Licensed Technology for the treatment of any and all veterinary and human medical conditions, including obesity, pain, inflammation and arthritis (the “2018 Field”).
Pursuant to the 2018 Hebrew Agreement, notwithstanding the grant of the 2018 Hebrew License, Yissum, on behalf of Hebrew University, will retain the right to (i) make, use and practice the 2018 Hebrew Licensed Technology for Hebrew University’s own research and educational purposes; (ii) license or otherwise convey to other academic and not-for-profit research organizations the 2018 Hebrew Licensed Technology for use in non-commercial research; and (iii) license or otherwise convey the 2018 Hebrew Licensed Technology to any third party for research or commercial applications outside the 2018 Field.
The 2018 Hebrew Agreement further provides that CBR Pharma is entitled to grant one or more sublicenses to the 2018 Hebrew Licensed Technology for exploitation in the 2018 Field.
12
All right, title and interest in and to the 2018 Hebrew Licensed Technology vest solely in Yissum, and CBR Pharma will hold and make use of the rights granted pursuant to the 2018 Hebrew License solely in accordance with the terms of the 2018 Hebrew Agreement.
As consideration for the 2018 Hebrew License, CBR Pharma paid Yissum a license fee of $75,000 and agreed to continue to pay an annual license maintenance fee (the “License Maintenance Fee”) of $50,000, beginning on May 1, 2019 and thereafter on the first day of May each year. The License Maintenance Fee is non-refundable, but may be credited each year against royalties on account of net sales of products made from May 1 to April 30 of each year.
Yissum has also agreed to undertake research and to synthesize chemical compounds that will be used by CBR Pharma, through additional research at both Oxford and Hebrew University, to develop orally active analgesic and anti-inflammatory medications. Compounds will be shipped from Hebrew University to Oxford for use in pre-clinical studies to establish efficacy in pain and inflammation.
Upon the achievement of certain milestones in respect of the chemical compounds derived from the 2018 Hebrew Licensed Technology, CBR Pharma is obligated to make certain payments to Yissum, including but not limited to the following:
| Milestone | Milestone Fee | |||
| Submission of the first IND testing for the FDA | $ | 75,000 | ||
| Commencement of one Phase 1/2 trial with the FDA | $ | 100,000 | ||
| Commencement of one Phase 3 trial with the FDA | $ | 150,000 | ||
| For each product market authorization/clearance (maximum of $500,000) | $ | 100,000 (maximum of $500,000) |
||
| For every $250 million in accumulated sales of the product until $1 billion in sales is achieved | $ | 250,000 | ||
CBR Pharma will pay Yissum royalties equal to (i) 3% of the net sales for the first annual $500 million of net sales, and (ii) 5% of the net sales after the net sales are at or in excess of $500 million.
In the event of a sale by CBR Pharma stockholders of their common shares or the transfer or assignment of the 2018 Hebrew Agreement, CBR Pharma is obligated to pay Yissum a fee of 5% of the consideration received by CBR Pharma pursuant to such corporate transaction. In the event of an initial public offering, or a go-public event, CBR Pharma was obligated to issue registered common shares to Yissum equal to 5% of the issued and outstanding common shares, on a fully-diluted basis, concurrently with the closing of such transaction. The Business Combination that was consummated on November 6, 2020, was considered a go-public event, pursuant to which the Company issued 240,541 of its common shares to Yissum prior to the closing of the Business Combination. See Note 12 - Commitments and Contingencies and Note 13 – Stockholders’ Equity of the financial statements for the fiscal period ended December 31, 2021 included herein for more information on the shares issued to Yissum as per the research and license agreement.
CBR Pharma has also agreed to reimburse Yissum (to a maximum of $30,000) for costs incurred for patent expenses.
Yissum and CBR Pharma also agreed to establish a research program for which CBR Pharma funded a $400,000 budget for the 12-month period ended May 2019, which is in the process of being extended by an amendment.
The 2018 Hebrew Agreement will terminate upon the occurrence of the later of the following: (i) the expiration of the last of the 2018 Hebrew Licensed Patents; (ii) the expiration of the last exclusivity on any product granted by any regulatory or government body; (iii) the expiration of a continuous period of twenty years during which there was no commercial sale of any product in any country; or (iv) if we elect to obtain an exclusive license to the know-how under the terms of the 2018 Hebrew Agreement, the expiration of such exclusive license.
13
On November 11, 2019, CBR Pharma entered into an additional research and license agreement (the “2019 Hebrew Agreement”) with Yissum, pursuant to which Yissum granted CBR Pharma a worldwide sole and exclusive license (the “2019 Hebrew License”) to develop and commercialize certain patents (the “2019 Hebrew Licensed Patents”), know-how and research results (collectively, the “2019 Hebrew Licensed Technology,” and together with the 2018 Hebrew Licensed Technology, the “Hebrew Licensed Technology”), in order to develop, manufacture, market, distribute, sell, repair and refurbish products, all within the use of the 2019 Hebrew Licensed Technology for (i) Cannabinoid phenolate metal salts, including mono, di and trivalent metals such as Li, Na, K, Ca, Mg, Zn, Fe and Al and their mixtures with native or synthetic cannabinoids, their pharmaceutical formulations, including for oral and topical administration; and (ii) pharmaceutical formulations, for the administration of cannabinoid chemical derivatives, including any and all veterinary and human medical conditions, including obesity, pain, inflammation and arthritis (the “2019 Field”).
Pursuant to the 2019 Hebrew Agreement, notwithstanding the grant of the 2019 Hebrew License, Yissum, on behalf of Hebrew University, will retain the right to (i) make, use and practice the 2019 Hebrew Licensed Technology for Hebrew University’s own research and educational purposes, but not for commercial purposes, and subject to the maintenance of confidentiality for any know-how or unpublished patent information contain in the 2019 Hebrew Licensed Technology; (ii) license or otherwise convey to other academic and not-for-profit research organizations the 2019 Hebrew Licensed Technology for use in non-commercial research and subject to the maintenance of confidentiality for any know-how or unpublished patent information contain in the 2019 Hebrew Licensed Technology; and (iii) license or otherwise convey the 2019 Hebrew Licensed Technology to any third party for research or commercial applications outside the 2019 Field, subject to the maintenance of confidentiality for any know-how or unpublished patent information contain in the 2019 Hebrew Licensed Technology.
The 2019 Hebrew Agreement further provides that CBR Pharma is entitled to grant one or more sublicenses to the 2019 Hebrew Licensed Technology for exploitation in the 2019 Field.
All right, title and interest in and to the 2019 Hebrew Licensed Technology vest solely in Yissum, and CBR Pharma will hold and make use of the rights granted pursuant to the 2019 Hebrew License solely in accordance with the terms of the 2019 Hebrew Agreement.
The 2019 Hebrew Licensed Technology will terminate upon the occurrence of the later of the following: (i) the expiration of the last of the 2019 Hebrew Licensed Patents; (ii) the expiration of the last exclusivity on any product granted by any regulatory or government body; (iii) the expiration of a continuous period of twenty years plus any applicable patent extension period, during which there was no commercial sale of any product in any country; or (iv) if we elect to obtain an exclusive license to the know-how under the terms of the 2019 Hebrew Agreement, the expiration of such exclusive license.
On January 1, 2020, CBR Pharma and Yissum entered into the first amendment to the 2018 Hebrew Agreement, which provided for additional research to be done at Yissum on new derivatives of certain molecules. Pursuant to the terms of the First Amendment, the Company will pay Yissum $200,000 per year plus 35% additional for University overhead for the additional research performed by each professor over an 18-month period, starting May 1, 2019. The additional research was initially expected to end in April 2021 and we are in negotiations with Yissum to extend the agreement.
Research Agreements with the University of Oxford
On November 1, 2013, our wholly-owned subsidiary 180 LP entered into an agreement (the “First Oxford Agreement”) with Oxford, pursuant to which 180 LP will sponsor Oxford’s research and development of repurposing anti-TNF for Dupuytren’s Contracture.
14
Pursuant to the First Oxford Agreement, each payment is to be made to ISIS Innovation (now Oxford University Innovation) at different milestones of the project, outlined below:
| Milestone | Milestone Fee | |||
| Minimum investment completed | £ | 10,000 | ||
| Initiation of Phase 2 trial for a licensed product | £ | 10,000 | ||
| Initiation of Phase 3 trial for a licensed product | £ | 10,000 | ||
| Registerable Phase 3 trial primary endpoint achieved for a licensed product | £ | 20,000 | ||
| Any issued U.S. patent of the licensed intellectual property rights | £ | 5,000 | ||
| Approval by FDA of an NDA filed by 180 LP or one of its sub-licensees for a licensed product | £ | 30,000 | ||
| Approval by EMA of an MAA filed by 180 LP or one of its sub-licensees for a licensed product | £ | 30,000 | ||
| First commercial sale of a licensed product by 180 LP or any sub-licensee in the U.S. | £ | 50,000 | ||
| First commercial sale of a licensed product by 180 LP or any sub-licensee in the EU | £ | 50,000 | ||
ISIS Innovation is also eligible for royalty payments equal to 0.5% of net sales in any country where there is a valid claim, 0.25% of net sales in other countries and a fee income royalty rate of 7.5% on all up-front, milestone and other one-off payments under or in connection with all sub-licenses and other contracts granted by 180 LP with respect to the licensed technology. The First Oxford Agreement is effective, unless earlier terminated, for so long as the specified patent application remains in effect as an issued patent, pending patent application or supplementary protection certificate or for a term of 20 years, whichever is longer.
On August 15, 2018, CannBioRex Pharma Limited, a company incorporated under the laws of England and Wales (“CannU.K.”) and a wholly-owned subsidiary of our wholly-owned subsidiary CBR Pharma, entered into the Research Agreement (the “Second Oxford Agreement”) with Oxford, pursuant to which CBR Pharma (through CannU.K.) has sponsored Oxford’s research and development of SCAs developed from the Hebrew Licensed Technology. At Oxford, the SCAs generated in the Hebrew University are being tested for analgesic and anti-inflammatory effects in established pre-clinical models.
Pursuant to the Second Oxford Agreement, Oxford undertook a research project (the “Research Project”) based around the clinical development of SCAs that are known to exhibit both anti-inflammatory and immunomodulatory properties. The aim of the Research Project was to develop and characterize chemical compounds that are synthesized at Hebrew University to create treatments for chronic pain, RA and other chronic inflammatory conditions, and to eventually obtain regulatory approval to initiate early-phase clinical trials by mid to late 2022 or as soon as possible thereafter. The Second Oxford Agreement had an initial term of one year beginning on March 22, 2019, but was extended by amendment to March 31, 2020, or any later date agreed to by the parties, unless terminated earlier. The Second Oxford Agreement was not extended any further after March 31, 2020, and CannU.K.’s relationship with Oxford continued with additional agreements with Oxford, as described below.
CannU.K., as the sponsor of the Research Project, made the following payments to Oxford pursuant to the Second Oxford Agreement:
| Milestone | Milestone Fee | |||
| Signature of the Oxford Agreement | £ | 166,800 | ||
| 6 months post start of the Research Project | £ | 166,800 | ||
| 9 months post start of the Research Project | £ | 166,800 | ||
| 12 months post start of the Research Project, after report | £ | 55,600 | ||
On September 18, 2020, CannU.K. entered into another research agreement with Oxford (the “Third Oxford Agreement”), pursuant to which CannU.K. sponsors work led by Prof. Nanchahal at the University of Oxford to investigate the mechanisms underlying fibrosis. In connection with the agreement, CannU.K. initially provided $100,000 and then at 6-month intervals further funding to support the salary of Dr. Lynn Williams and consumables.
CannU.K., as the sponsor, agreed to make the following payments to Oxford pursuant to the Third Oxford Agreement:
| Milestone | Amount Due (excluding VAT) | |||
| 30 days post signing of the Third Oxford Agreement | £ | 80,000 | ||
| 6 months post signing of the Third Oxford Agreement | £ | 178,867 | ||
| 12 months post signing of the Third Oxford Agreement | £ | 178,867 | ||
| 24 months post signing of the Third Oxford Agreement | £ | 178,867 | ||
| 36 months post signing of the Third Oxford Agreement | £ | 178,867 | ||
On September 21, 2020, CannU.K. entered into another research agreement with Oxford (the “Fourth Oxford Agreement”), pursuant to which CannU.K. agreed to sponsor work at the University of Oxford to develop and characterize novel cannabinoid derived new chemical entities (NCEs) for the treatment of inflammatory diseases towards initiation of early phase clinical trials in patients within a period of 3 years.
15
CannU.K., as the sponsor, agreed to make the following payments to Oxford pursuant to the Fourth Oxford Agreement:
| Milestone | Amount Due (excluding VAT) | |||
| 30 days post signing of the Fourth Oxford Agreement | £ | 101,778 | ||
| 6 months post signing of the Fourth Oxford Agreement | £ | 101,778 | ||
| 12 months post signing of the Fourth Oxford Agreement | £ | 101,778 | ||
| 18 months post signing of the Fourth Oxford Agreement | £ | 101,778 | ||
| 24 months post signing of the Fourth Oxford Agreement | £ | 101,778 | ||
On March 22, 2022, CannU.K. entered into an amendment to the Fourth Oxford Agreement, to extend the research period to December 31, 2023, at no additional cost to CannU.K.
On May 24, 2021, CannU.K. entered into another research agreement with Oxford (the “Fifth Oxford Agreement”), pursuant to which CannU.K. will sponsor work at the University of Oxford to conduct a multi-center, randomized, double blind, parallel group, feasibility study of anti-TNF injection for the treatment of adults with frozen shoulder during the pain-predominant phase.
CannU.K., as the sponsor, agreed to make the following payments to Oxford pursuant to the Fifth Oxford Agreement:
| Milestone | Amount Due (excluding VAT) | |||
| Upon signing of the Fifth Oxford Agreement | £ | 70,546 | ||
| 6 months post signing of the Fifth Oxford Agreement | £ | 70,546 | ||
| 12 months post signing of the Fifth Oxford Agreement | £ | 70,546 | ||
| 24 months post signing of the Fifth Oxford Agreement | £ | 70,546 | ||
16
Oxford License Agreement
On November 3, 2021, we entered into an exclusive license agreement with Oxford University Innovation Limited (“Oxford License Agreement”), pursuant to which we were granted the rights to certain patents related to the HMGB1 molecule for liver regeneration.
Pursuant to the Oxford License Agreement, the Company agreed to the following payment terms:
| Payment | Amount Due | |||
| Past patent costs | £ | 49,207 | ||
| License fee | £ | 10,000 | ||
| Annual maintenance fee | £ | 3,000 | ||
| Milestone | Amount Due | |||
| Submission of IND | £ | 25,000 | ||
| 1st Subject dosed in Phase I studies for each product, each indication | £ | 25,000 | ||
| 1st Subject dosed in Phase II studies for each product, each indication | £ | 100,000 | ||
| 1st Subject dosed in Phase III studies for each product, each indication | £ | 50,000 | ||
| Submission of New Drug Application for each product for each indication | £ | 50,000 | ||
| Issued US patent, per patent | £ | 5,000 | ||
| Receipt of Regulatory Approval in the US for each product for each indication | £ | 1,250,000 | ||
| Receipt of Regulatory Approval in the EU or U.K. for each product for each indication | £ | 550,000 | ||
| Receipt of Regulatory Approval in the Japan for each product for each indication | £ | 150,000 | ||
| Aggregate Net Sales Exceed $5Bn | £ | 10,000,000 | ||
| Aggregate Net Sales Exceed $10Bn | £ | 50,000,000 | ||
| Net Sales (US$) | Royalty Rate | |||
| < $250M | 1.00 | % | ||
| $250M - $1B | 2.00 | % | ||
| $1B - $10B | 3.00 | % | ||
| > $10B | 3.50 | % | ||
Stanford License Agreement
On May 8, 2018, Katexco Pharmaceuticals Corp, a wholly-owned subsidiary of our wholly-owned subsidiary Katexco, entered into an option agreement (the “Stanford Option”) with the Board of Trustees of the Leland Stanford Junior University (“Stanford”), pursuant to which Stanford granted Katexco an option to acquire an exclusive license for the development and commercialization of certain inventions. In consideration for the Stanford Option, Katexco paid Stanford $10,000 (the “Option Payment”), creditable against the license issue fee agreement.
On July 25, 2018 (the “Stanford Effective Date”) Katexco exercised the Stanford Option, and entered into an exclusive license agreement (the “Stanford License Agreement”) with Stanford, pursuant to which Katexco was granted the rights to certain U.S. patents related to (i) alpha B-crystallin as a therapy for autoimmune demyelination and (ii) peptides as short as six amino acids that form amyloid fibrils that activate B-1 cells and macrophages and are anti-inflammatory and therapeutic in autoimmune and neurodegenerative diseases (the “Stanford Licensed Patents”). Through the Stanford License Agreement, Katexco established research facilities at Stanford. We will support the clinical development of the lead compound(s), culminating in Phase 1 and Phase 2 clinical trials to establish potential clinical utility in ulcerative colitis indications.
Under the Stanford License Agreement, no rights of Stanford, including intellectual property rights, are granted to Katexco other than those rights granted under the Stanford Licensed Patents.
As consideration for the grant of the Stanford Licensed Patents, Katexco paid Stanford an initial fee of $50,000, inclusive of the Option Payment. The Company also issued 111,466 common shares to Stanford, and provided a letter stating the value of such shares. A portion of the shares issued to Stanford were later distributed to five individuals, including our Chief Scientific Officer and co-chairman.
17
Beginning upon the first anniversary of the Stanford Effective Date and each anniversary thereafter, Katexco will pay Stanford an annual license maintenance fee of $20,000 on the first and second anniversaries and $40,000 on each subsequent anniversary. Furthermore, Katexco is obligated to make the following payments, including (i) $100,000 upon initiation of Phase 2 trial, (ii) $500,000 upon the first FDA approval of a product (the “Licensed Product”) resulting from the Stanford Licensed Patents, and (iii) $250,000 upon each new Licensed Product thereafter. Royalties, calculated at 2.5% of net sales (calculated as gross revenue received by Katexco or its sublicensees, their distributors or designees, from the sale, transfer or other disposition of products based on the Stanford Licensed Patents minus 5%), will be payable to Stanford. In addition, Katexco has reimbursed Stanford $51,385 to offset the Stanford Licensed Patent’s patenting expenses, and will reimburse Stanford for all Stanford Licensed Patent’s patenting expenses, including any interference and or re-examination matters, incurred by Stanford after March 3, 2018.
We can terminate the Stanford License Agreement without cause by providing a 30-day notice. In the case of a change of control, upon the assignment of the Stanford License Agreement, Katexco is obligated to pay Stanford a $200,000 change of control fee. The Stanford License Agreement also provides Stanford with the right to purchase for cash up to either (i) 10% or (ii) the percentage necessary for Stanford to maintain its pro rata ownership interest in Katexco, of Katexco’s equity securities issued in a private offering. The shares issued to Stanford in connection with the Stanford License Agreement, gave Stanford and the five individuals who received a portion of the shares a total ownership of 2.11% in Katexco’s stock, prior to the July 2019, corporate restructuring completed between 180 and each of 180 LP, Katexco and CBR Pharma, pursuant to which 180 LP, Katexco and CBR Pharma became wholly-owned subsidiaries of 180LS (the “Reorganization”) under the Business Corporations Act (British Columbia).
The Evotec Agreement
On June 7, 2018, our wholly-owned subsidiary Katexco entered into the Evotec Agreement with Evotec, a leading CRO, pursuant to which Evotec was retained to perform certain research services. Pursuant to the Evotec Agreement, the goal of the joint project (the “Evotec Project”) is to identify small molecules that pharmacologically stimulate the human ChrFam7a receptor and function. The Evotec Project is being conducted in two phases over a 24-month period where resources are allocated by the steering committee, which is controlled equally by the parties to the Evotec Agreement, on a quarterly basis.
Subject to certain exemptions described in the Evotec Agreement, Katexco owns all intellectual property rights, conceived, invented, discovered or made by Evotec during the performance of its services, other than intellectual property rights owned or controlled by Evotec relating to its already existing technology and components to be used in the services to be provided under the Evotec Agreement.
The Evotec Agreement is subject to a minimum payment of $4,937,500 and a maximum payment of $5,350,250 to Evotec. This program was paused in mid-2019 and the Company is currently in negotiations with Evotec to continue this program.
The Petcanna Agreement
On August 20, 2018, we entered into a sublicense agreement with Petcanna Pharma Corp. (“Petcanna”), a private company which was founded by Prof. Sir Marc Feldmann (our Co-Executive Chairman), and Yissum (the “Petcanna Agreement”).
Under the Petcanna Agreement, we granted Petcanna an exclusive, worldwide, non-transferable, non-sublicensable sublicense to make commercial use of the certain patents related to cyclohexenyl compounds and listed in the Petcanna Agreement (the “Petcanna IP”) in order to develop, manufacture, market, distribute or sell products that incorporate the Petcanna IP in products that are intended for the treatment of veterinary medical conditions, initially osteoarthritis.
As consideration for the sublicense, Petcanna agreed to issue to us approximately 9,000,000 of Petcanna’s common shares in the fourth quarter of 2018. As of the date of this filing, Petcanna has not issued shares to any shareholder and has not commenced operations. We intend to retain 85% of such shares, and transfer 15% of such shares to Yissum. In the event that Yissum does not accept such shares, we will have an obligation to pay Yissum 15% of the-then current fair market value of such shares. Petcanna will also pay a 1% royalty to us on Petcanna’s net sales of products that incorporate the Petcanna IP.
18
All right, title and interest in and to the Petcanna IP, including any improvements to the Petcanna IP, will vest solely in our company.
Unless the parties to the Petcanna Agreement agree otherwise in writing, the Petcanna Agreement will terminate on the occurrence of the later of: (i) the date of expiration of the last of the Petcanna IP, (ii) the date of the final expiration of exclusivity on any Product granted by any regulatory or government body, and (iii) the expiration of a continuous period of twenty (20) years during which there was no First Commercial Sale of any product. The terms “Product” and “First Commercial Sale” apply as they are defined in the Petcanna Agreement. Our ability to grant this sublicense to Petcanna is contingent upon (i) Yissum having the necessary rights to the Hebrew Patent Applications assigned to it from all applicable parties, (ii) Yissum being able to grant a license to us per the terms of the Hebrew Agreement, and (iii) the Hebrew Patent Applications and any related resulting patents being valid and maintained in good standing for the respective terms of the Hebrew Licensing Agreement and the Petcanna Agreement.
Kennedy License Agreement
On September 27, 2019, our wholly-owned subsidiary 180 LP entered into an exclusive license agreement (the “Kennedy License Agreement”) with the Kennedy Trust For Rheumatology Research (“Kennedy”), pursuant to which Kennedy granted to 180 LP an exclusive license in the U.S., Japan and member countries of the EU (including the United Kingdom), to certain licensed patents (the “Kennedy Licensed Patents”), including the right to grant sublicenses, and the right to research, develop, sell or manufacture any pharmaceutical product (i) whose research, development, manufacture, use, importation or sale would infringe on the Kennedy Licensed Patents absent the license granted under the Kennedy License Agreement or (ii) containing an antibody that is a fragment of or derived from an antibody whose research, development, manufacture, use, importation or sale would infringe on the Kennedy Licensed Patents absent the license granted under the Kennedy License Agreement, for all human uses, including the diagnosis, prophylaxis and treatment of diseases and conditions.
Under the Kennedy License Agreement, Kennedy reserves the perpetual, irrevocable, non-exclusive, royalty-free, sublicensable, worldwide right for the Kennedy Licensed Patents and its affiliates, employees, students and other researchers to carry out any acts which would otherwise infringe on the Kennedy Licensed Patents for the purposes of teaching and carrying out research and development, including the right to accept external sponsorship for such research and development and the right to grant sub-licenses for the same purposes.
As consideration for the grant of the Kennedy Licensed Patents, 180 LP paid Kennedy an upfront fee of £60,000, and will also pay Kennedy royalties equal to (i) 1% of the net sales for the first annual $1 billion of net sales, and (ii) 2% of the net sales after the net sales are at or in excess of $1 billion, as well as 25% of all sublicense revenue, provided that the amount of such percentage of sublicense revenue based on amounts which constitute royalties shall not be less than 1% on the first cumulative $1 billion of net sales of the products sold by such sublicenses or their affiliates, and 2% on that portion of the cumulative net sales of the products sold by such sublicenses or their affiliates in excess of $1 billion.
The term of the royalties paid to Kennedy will expire on the later of (i) the last valid claim of a patent included in the Kennedy Licensed Patents which covers or claims the exploitation of a product in the applicable country; (ii) the expiration of regulatory exclusivity for the product in the country; or (iii) 10 years from first commercial sale of the product in the country.
We may terminate the Kennedy License Agreement without cause by providing 90-days’ notice.
19
Consulting Agreements
The Consulting Agreements are each described below.
Inflammation consultancy Agreements with each of Prof. Sir Marc Feldmann and Prof. Jagdeep Nanchahal
On November 1, 2013, our wholly-owned subsidiary 180 LP entered into letter agreements regarding inflammation consultancy services (each, an “Inflammation Consultancy Agreement”, and, collectively, the “Inflammation Consultancy Agreements”) with Isis Innovation Limited for the services of each of Prof. Sir Marc Feldmann and Prof. Jagdeep Nanchahal (each, an “Inflammation Consultant”). Pursuant to the Inflammation Consultancy Agreements, each Inflammation Consultant agreed to provide advice and expertise on inflammatory and degenerative diseases including fibrosis as exemplified by Dupuytren’s Contracture and osteoinduction (bone formation), in relation to the technology, programs and products of 180 LP, and, specifically, to provide general and specific advice and guidance on how 180 LP might further develop its different programs that are ongoing, contemplated, or conceived at or by 180 LP (the “Inflammation Consulting Services”).
In consideration of the Inflammation Consulting Services, Prof. Sir Marc Feldmann and Prof. Jagdeep Nanchahal were paid a fixed fee of $500 and $10,000 per annum, respectively.
The initial term of each Advisory Services Agreement was until November 1, 2015. On November 8, 2015, each of the Advisory Services Agreement was extended until November 1, 2020. A new contract with Prof. Jagdeep Nanchahal is described below. Prof. Sir Marc Feldmann has an employment agreement with CannU.K. which is described below.
Prof . Jagdeep Nanchahal Consulting Agreement
On February 25, 2021, we (and CannBioRex Pharma Limited, which was added as a party to the agreement later), entered into a Consultancy Agreement dated February 22, 2021, and effective December 1, 2020, with Prof. Jagdeep Nanchahal (as amended, the “Consulting Agreement”). Prof. Nanchahal has been providing services to the Company and/or its subsidiaries since 2014, and is currently a greater than 5% stockholder of the Company and the Chairman of our Clinical Advisory Board.
On March 31, 2021, we entered into a First Amendment to Consultancy Agreement with Prof. Jagdeep Nanchahal, which amended the Consultancy Agreement entered into with Prof. Nanchahal on February 25, 2021, to include CannBioRex Pharma Limited, a corporation incorporated and registered in England and Wales (“CannBioRex”), and an indirect wholly-owned subsidiary of the Company, as a party thereto, and to update the prior Consultancy Agreement to provide for cash payments due to Prof. Nanchahal to be paid by CannBioRex, for tax purposes, provide for CannBioRex to be party to certain other provisions of the agreement and to provide for the timing of certain cash bonuses due under the terms of the agreement.
Prof. Nanchahal is a surgeon scientist focusing on defining the molecular mechanisms of common diseases and translating his findings through to early phase clinical trials. He undertook his PhD, funded by the U.K. Medical Research Council, whilst a medical student in London and led a lab group funded by external grants throughout his surgical training. After completing fellowships in microsurgery and hand surgery in the USA and Australia, he was appointed as a senior lecturer at Imperial College. His research is focused on promoting tissue regeneration by targeting endogenous stem cells and reducing fibrosis. In 2013 his group identified anti-tumor necrosis factor (TNF) as therapeutic target for Dupuytren’s Contracture, a common fibrotic condition of the hand. He is currently leading a Phase 2b clinical trial funded by the Wellcome Trust and Department of Health to assess the efficacy of local administration of anti-TNF in patients with early-stage Dupuytren’s Contracture and a clinical trial for patients with early-stage frozen shoulder. He is a proponent of evidence-based medicine and was the only plastic surgery member of the NICE Guidance Development Groups on complex and non-complex fractures. He was a member of the group that wrote the Standards for the Management of Open Fractures published in 2020. This is an open-source publication to facilitate the care of patients with these severe injuries.
Pursuant to the Consulting Agreement, Prof. Nanchahal agreed, during the term of the agreement, to serve as a consultant to the Company and provide such services as the Chief Executive Officer and/or the board of directors of the Company shall request from time to time, including but not be limited to: (1) conducting clinical trials in the fields of Dupuytren’s Contracture, frozen shoulder and post-operative delirium/cognitive decline; and (2) conducting laboratory research in other fibrotic disorders, including fibrosis of the liver and lung (collectively, the “Services”).
20
In consideration for providing the Services, the Company (through CannBioRex Pharma Limited) agreed to pay Prof. Nanchahal 15,000 British Pounds (GBP) per month (approximately $20,800) during the term of the agreement, increasing to GBP 23,000 (approximately $32,000) on the date (a) of publication of the data from the phase 2b clinical trial for Dupuytren’s Contracture (RIDD) and (b) the date that the Company has successfully raised over $15 million in capital. The fee will increase annually thereafter to reflect progression in other clinical trials and laboratory research as approved by the board of directors. The Company also agreed to pay Prof. Nanchahal a bonus (“Bonus 1”) in the sum of GBP 100,000 upon submission of the Dupuytren’s Contracture clinical trial data for publication in a peer-reviewed journal, which submission occurred in December 2021, and which bonus was paid in December 2021. In addition, for prior work performed, including completion of the recruitment to the RIDD (Dupuytren’s) trial, the Company agreed to pay Prof. Nanchahal GBP 434,673 (approximately $605,000) (“Bonus 2”). At the election of Prof. Nanchahal, Bonus 2 shall be paid at least 50% (fifty percent) or more, as Prof. Nanchahal elects, in shares of the Company’s common stock, at a share price of $3.00 per share, or the share price on the date of the grant, whichever is lower, with the remainder paid in GBP. Bonus 2 shall be deemed earned and payable upon the Company raising a minimum of $15 million in additional funding, through the sale of debt or equity, after December 1, 2020 (the “Vesting Date”) and shall not be accrued, due or payable prior to such Vesting Date. Bonus 2 shall be payable by the Company within 30 calendar days of the Vesting Date. Finally, Prof. Nanchahal shall receive another one-time bonus (“Bonus 3”) of GBP 5,000 (approximately $7,000) on enrollment of the first patient to the phase 2 frozen shoulder trial, and another one-time bonus (“Bonus 4”) of GBP 5,000 (approximately $7,000) for enrollment of the first patient to the phase 2 delirium/POCD trial. On March 30, 2021, the Company issued Prof. Nanchahal 100,699 shares of Company common stock in lieu of GBP 217,337 and on April 15, 2021, the Company issued Prof. Nanchahal 37,715 shares of Company common stock in lieu of GBP 82,588. The Company also waived the requirement for the Company having to raise $15 million in order for Prof. Nanchahal to agree to receive an aggregate of GBP 300,000 via the issuance of shares. Prof. Nanchahal agreed that the remaining GBP 134,673 that is due pursuant to Bonus 2 shall be paid after the Company has raised a minimum of $15 million in additional funding. On August 23, 2021, at the request of Prof. Nanchahal, the Company agreed to issue Prof. Nanchahal 61,535 shares of common stock in consideration for the remaining 31% (or 134,748.63 GBP, or $184,605.62) of Bonus 2, based on a $3.00 per share price. The shares were issued under the Company’s 2020 Omnibus Incentive Plan, which has been approved by stockholders.
Notwithstanding the above, the board of directors or Compensation Committee of the Company may grant Prof. Nanchahal additional bonuses from time to time in their discretion, in cash, stock or options.
The Consulting Agreement has an initial term of three years, and renews thereafter for additional three-year terms, until terminated as provided in the agreement. The Consulting Agreement can be terminated by either party with 12 months prior written notice (provided the Company’s right to terminate the agreement may only be exercised if Prof. Nanchahal fails to perform his required duties under the Consulting Agreement), or by the Company immediately if (a) Prof. Nanchahal fails or neglects efficiently and diligently to perform the Services or is guilty of any breach of its or his obligations under the agreement (including any consent granted under it); (b) Prof. Nanchahal is guilty of any fraud or dishonesty or acts in a manner (whether in the performance of the Services or otherwise) which, in the reasonable opinion of the Company, has brought or is likely to bring Prof. Nanchahal, the Company or any of its affiliates into disrepute or is convicted of an arrestable offence (other than a road traffic offence for which a non-custodial penalty is imposed); or (c) Prof. Nanchahal becomes bankrupt or makes any arrangement or composition with his creditors. If the Consulting Agreement is terminated by the Company for any reason other than cause, Prof. Nanchahal is entitled to a lump sum payment of 12 months of his fee as at the date of termination.
The Consulting Agreement includes a 12 month non-compete and non-solicitation obligation of Prof. Nanchahal, preventing him from competing against the Company in any part of any country in which he was actively engaged in the Company’s business, subject to certain exceptions, including research conducted at the University of Oxford. The Consulting Agreement also includes customary confidentiality and assignment of inventions provisions, in each case subject to the Company’s previously existing agreements with various universities, including the University of Oxford, where Prof. Nanchahal serves as a Professor of Hand, Plastic and Reconstructive Surgery.
21
Service Agreement with Prof. Sir Marc Feldmann
On June 1, 2018, CannBioRex Pharma Limited (“CannBioRex”) and Prof. Sir Marc Feldmann Ph.D., our Executive Co-Chairman, entered into a Service Agreement (the “Feldmann Employment Agreement”). Pursuant to the Feldmann Employment Agreement, Prof. Sir Feldmann serves as the Chairman, CEO and Executive Director of CannBioRex or in such other capacity consistent with his status. Prof. Sir Feldmann’s responsibilities include those customary for the roles in which he serves. Prof. Sir Feldmann receives compensation of £115,000 per year, with annual compensation reviewed by the Board and eligibility for discretionary bonuses, as determined by the Board. CannBioRex also reimburses Prof. Sir Feldmann’s travelling and other business expenses.
Pursuant to the Feldmann Employment Agreement, all intellectual property rights created by Prof. Sir Feldmann or related to his employment belong to and vest in CannBioRex.
The Feldmann Employment Agreement contains a customary non-compete clause prohibiting Prof. Sir Feldmann from working for any competing businesses during the term of his employment, or holding equity in other businesses, except he may hold or beneficially own securities of publicly-traded companies if the aggregate beneficial interests of him and his family does not exceed 5% of that class of securities.
Prof. Sir Feldmann is also prohibited for 12 months following termination (the “Post-Termination Period”) be involved in any capacity with a competing business or potential joint venturer in the United Kingdom or in any other country. During the Post-Termination Period, he may not solicit business from CannBioRex and its affiliates’ customers; or any company with whom he was activity involved in the course of his employment; or about which he holds confidential information. Prof. Sir Feldmann further covenants to not interfere with CannBioRex’s business relationships by inducing or attempting to induce suppliers to take adverse actions during the Post-Termination Period. He also agrees not to induce or attempt to induce any CannBioRex employee to leave the company during the Post-Termination Period. The Feldmann Employment Agreement contains customary non-disclosure and confidentiality obligations, sick leave and vacation time.
The Feldmann Employment Agreement does not have a fixed term. Either party may terminate the agreement by delivering written notice 9 months in advance. CannBioRex may also terminate the Feldmann Employment Agreement at any time with immediate effect by giving written notice. If CannBioRex terminates Prof. Sir Feldmann’s employment without providing 9 months written notice, he will become entitled to a payment equal to his basic salary he would have been entitled to receive if 9 months’ notice were given. The governing law for the Feldmann Employment Agreement is the law of England.
The Board of Directors, as recommended by the Compensation Committee of the Company (and/or the Compensation Committee) or separately, may also award Prof. Sir Feldmann bonuses from time to time (in stock, options, cash, or other forms of consideration) in its discretion.
On November 17, 2021, the Board of Directors, as recommended by the Compensation Committee, increased the salary of Prof. Sir Feldmann to $225,000 per annum.
On December 8, 2021, Prof. Sir Feldmann was granted stock options to purchase 25,000 shares of the Company’s common stock, which have a term of 10 years; an exercise price equal to the fair market value of the Company’s common stock on the date of grant, $3.95 per share, and are subject to the Company’s 2020 Omnibus Incentive Plan.
22
Consultancy Agreement with Prof. Lawrence Steinman
On May 31, 2018, CannBioRex Pharma Limited (“CannBioRex”) and Prof. Lawrence Steinman, our Executive Co-Chairman, entered into a Consultancy Agreement (the “Steinman Agreement”). Prof. Steinman committed to making himself available to provide services to CannBioRex that it may require of Prof. Steinman’s expertise. In consideration for the services provided, CannBioRex will pay Prof. Steinman approximately $50,000 per year, in monthly installments. Prof. Steinman is also entitled to reimbursement of all business expenses, including travel.
Although Prof. Steinman is not prohibited from engaging in other business activities during the term of the agreement, he must remain compliant with the terms of the agreement and seek prior written consent before conducting business with a business similar to, or competitive with, CannBioRex. The Steinman Agreement contains customary confidentiality and assignment of intellectual property provisions.
The Steinman Agreement had a two-year term. The agreement may be terminated earlier by Prof. Steinman or CannBioRex with six months’ written notice. CannBioRex may terminate the agreement at any time with immediate effect if Prof. Steinman fails or neglects to efficiently and diligently perform services pursuant to the agreement or breaches its terms; is guilty of fraud, dishonesty or acts in a manner which the Company reasonably deems is likely to disparage himself or the Company; becomes unable to provide services for ten working days in any month; or Prof. Steinman becomes bankrupt or arranges for compromises with his creditors. Upon termination of the Steinman Agreement, Prof. Steinman must return property related to the consultancy and delete his own electronic records. The parties have continued to operate under the terms of the agreement even though the agreement expired.
In addition, Prof. Steinman had a verbal agreement with Katexco Pharmaceuticals Corp., a wholly-owned subsidiary of the Company to provide services for $100,000 per year, since Katexco’s incorporation in 2018.
Both of these agreements have been terminated and replaced with a new consulting agreement with 180 Life Sciences, as described in detail below.
Lawrence Steinman, M.D. Consulting Agreement
On November 17, 2021, and effective on November 1, 2021, the Company entered into a Consulting Agreement with Lawrence Steinman, M.D., the Company’s Executive Co-Chairman (the “Consulting Agreement”). Pursuant to the Consulting Agreement, Dr. Steinman agreed to provide certain consulting services to the Company, including, but not limited to, participating in defining and setting strategic objectives of the Company; actively seeking out acquisition and merger candidates; and having primary scientific responsibility for the Company’s á7nAChR platform (collectively, the “Services”). The term of the agreement is for one year (the “Initial Term”); provided that the agreement automatically extends for additional one year periods after the Initial Term (each an “Automatic Renewal Term” and the Initial Term together with all Automatic Renewal Terms, if any, the “Term”), subject to the Renewal Requirements (described below), in the event that neither party provided the other written notice of their intent not to automatically extend the term of the agreement at least 30 days prior to the end of the Initial Term or any Automatic Renewal Term. The Term can only be extended for an Automatic Renewal Term, provided that (i) Dr. Steinman is re-elected to the Board of Directors (the “Board”) at the Annual Meeting of Stockholders of the Company immediately preceding the date that such Automatic Renewal Term begins; (ii) the Board affirms his appointment as Co-Chairman for the applicable Automatic Renewal Term (or fails to appoint someone else as Co-Chairman prior to such applicable Automatic Renewal Term) and (iii) Dr. Steinman is continuing in his role of having the responsibility for the scientific development for the Company’s á7nAChR platform (the “Renewal Requirements”). The Consulting Agreement also expires immediately upon the earlier of: (i) the date upon which Dr. Steinman no longer serves as Co-Chairman and no longer has primary scientific responsibility for our á7nAChR platform; and (ii) any earlier date requested by either (1) the Company (as evidenced by a vote of a majority of the Board (excluding Dr. Steinman) at a meeting of the Board), or (2) Dr. Steinman (as evidenced by written notice from Dr. Steinman to the Board). Additionally, the Company may terminate the Consulting Agreement immediately and without prior notice if Dr. Steinman is unable or refuses to perform the Services, and either party may terminate the Consulting Agreement immediately and without prior notice if the other party is in breach of any material provision of the Consulting Agreement.
23
The Company agreed to pay Dr. Steinman $225,000 per year during the term of the agreement, along with a one-time payment of $43,750, representing the difference between his old compensation and new compensation, dating back to April 1, 2021. Pursuant to the Consulting Agreement, Dr. Steinman agreed to not compete against the Company, unless approved in writing by the Board of Directors, during the term of the agreement, and also agreed to certain customary confidentiality provisions and assignment of inventions requirements. The Consulting Agreement also has a 12 month non-solicitation prohibition following its termination.
On December 8, 2021, Dr. Steinman was also granted stock options to purchase 25,000 shares of the Company’s common stock, which have a term of 10 years; an exercise price equal to the fair market value of the Company’s common stock on the date of grant, $3.95 per share, and are subject to the Company’s 2020 Omnibus Incentive Plan. In addition, beginning in calendar year 2022, for each year during the Term of the Consulting Agreement, the Company will, subject to future approval by the Board, grant Dr. Steinman $125,000 of value of equity compensation. Future equity grants will vest over a 48 month period and be in accordance with the Plan. Timing of the future grants, nature of the equity grants (e.g., RSU, PSU, restricted stock, etc.) and any changes in the value of future equity will be recommended by the Company’s Compensation Committee and/or Audit Committee and approved by the Board.
Intellectual Property
Our success depends in significant part on our ability to protect the proprietary elements of our product candidates, technology and know-how, to operate without infringing on the proprietary rights of others, and to defend challenges and oppositions from others and prevent others from infringing on our proprietary rights. We have sought, and will continue to seek, patent protection in the U.S., U.K., Europe and other countries for our proprietary technologies. Our intellectual property portfolio as of March 28, 2022, includes sixteen patent families with issued and/or pending claims, pharmaceutical formulations, drug delivery and the therapeutic uses of SCAs, as well as know-how and trade secrets, when including patents held by our partners of which we have exclusive rights.
Within the U.S., we and/or our partners have licensed eight issued patents and thirteen pending patent applications under active prosecution. Outside of the U.S., assuming the E.U. as a single jurisdiction, there are an additional twelve issued patents and 23 pending patent applications under active prosecution. Our policy is to seek patent protection for the technology, inventions and improvements that we consider important to the development of our business, but only in those cases where we believe that the costs of obtaining patent protection is justified by the commercial potential of the technology, and typically only in those jurisdictions that we believe present significant commercial opportunities. We also rely on trademarks, trade secrets, know-how and continuing innovation to develop and maintain our competitive position.
The term of individual patents depends upon the countries in which they are obtained. In most countries in which we have filed, the patent term is 20 years from the earliest date of filing a non-provisional patent application. In the U.S., a patent’s term may be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the U.S. Patent and Trademark Office (“USPTO”), in granting a patent, or may be shortened if a patent is terminally disclaimed over another patent.
The term of a patent that covers an FDA-approved drug may also be eligible for extension, which permits term restoration as compensation for the term lost during the FDA regulatory review process. The Drug Price Competition and Patent Term Restoration Act of 1984 (the Hatch-Waxman Act) permits an extension of up to five years beyond the expiration of the patent. The length of the patent term extension is related to the length of time the drug is under regulatory review. Extensions cannot extend the remaining term of a patent beyond 14 years from the date of product approval and only one patent applicable to an approved drug may be extended. Similar provisions to extend the term of a patent that covers an approved drug are available in Europe and other non-U.S. jurisdictions.
24
To protect our rights to any of our issued patents and proprietary information, we may need to litigate against infringing third parties, avail ourselves of the courts or participate in hearings to determine the scope and validity of those patents or other proprietary rights.
We also rely on trade secret protection for our confidential and proprietary information. Our policy requires our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors to execute confidentiality agreements upon the commencement of employment or consulting relationships with us.
From time to time, in the normal course of our operations, we will be a party to litigation and other dispute matters and claims relating to intellectual property.
180LS’ Research, Development and License Agreements
180LS has entered into research and licensing agreements with various parties, including the Hebrew University of Jerusalem and Oxford. For information regarding these agreements, see “Material Agreements”, above.
Competition
Below is a description of the competitive environment of each of our product candidate development platforms and potential product candidates.
Dupuytren’s Contracture
Our treatment is for early-stage Dupuytren’s Contracture, for which, to our knowledge, there is no approved treatment. Existing treatments focus on late stage Dupuytren’s Contracture, when the fingers are irreversibly curled into the palm. Surgery remains the typical standard treatment but the relatively long post-operative rehabilitation has driven the reach for less invasive techniques. Xiaflex, a drug developed by Auxilium, has shown effective in treating patients with developed contractures although many patients experience relatively mild side effects. An alternative approach is disruption of the late-stage cords with a needle and data from a comparative clinical trial published in the Journal of Bone and Joint Surgery (American) in 2018 showed similar recurrence rates between collagenase and percutaneous needle fasciotomy at 2 years. A clinical trial funded by the National Institute for Health Research Health Technology Assessment Programme (U.K.) is currently underway in the U.K., comparing the cost efficacy of surgery for Dupuytren’s Contracture with collagenase treatment. The aims of the study are to determine (i) whether collagenase injections are as effective and as safe as surgery for treating this condition and (ii) the costs of both treatments.
SCAs
Despite roughly 3,000 pharmaceutical and biotechnology companies globally, only a handful of companies are actively developing synthetic cannabinoids for human and veterinary health. Presently, most of the focus of these companies is on pain management, multiple sclerosis and epileptic-seizure disorders, and most of these companies are using natural plant products.
We expect that the market for cannabinoid therapeutics will also grow significantly in the coming years due to increasing awareness of the potential benefits that cannabinoid products may provide over existing therapies. Interest in cannabinoid therapeutics has increased over the past several years as some pre-clinical and clinical data has emerged highlighting the potential efficacy and safety benefits of cannabinoid and synthetic cannabinoid therapeutics. Pharmaceutical companies that have publicized their engagement in testing cannabinoid and synthetic cannabinoid therapeutics include Zynerba, Skye Bioscience, IntelGenx, Ananda Scientific, InMed Pharmaceuticals, GW Pharmaceuticals PLC (acquired by Jazz Pharmaceuticals), Tetra Bio-Pharma, and GB Sciences.
25
Multiple companies are working in the cannabis therapeutic area and are pursuing regulatory approval for their product candidates. For example, GW Pharmaceuticals PLC, which markets Sativex, a botanical cannabinoid oral mucosal for the treatment of spasticity due to multiple sclerosis is seeking FDA approval in the U.S., and Epidiolex, a liquid formulation of highly purified cannabidiol extract, which was recently approved as a treatment for Dravet’s Syndrome, Lennox Gastaut Syndrome, and various childhood epilepsy syndromes. Skye Bioscience, Inc. is focused on the discovery, development and commercialization of synthetic cannabinoid derived therapeutics to target glaucoma. Corbus Pharmaceuticals Holdings, Inc.’s lead drug candidate, lenabasum, is a novel, synthetic, oral, cannabinoid (CB2 agonist) designed to treat four serious and rare chronic inflammatory diseases (systemic sclerosis (“SSc”), dermatomyositis (“DM”), cystic fibrosis (“CF”) and systemic lupus erythematosus), and the FDA has granted lenabasum Orphan Drug Designation as well as Fast Track Status for SSc and CF, and Orphan Drug Designation for DM. Zynerba Pharmaceuticals focuses on pharmaceutically-produced transdermal cannabinoid therapies for rare and near-rare neuropsychiatric disorders and is currently evaluating ZygelTM, a patent-protected transdermal CBD gel for the treatment of Fragile X syndrome, for which it filed an NDA with the FDA, developmental and epileptic encephalopathies and Autism Spectrum Disorder.
a7nAChR
Competition to the acetylcholine receptor program is diverse, ranging from a small biotechnology company, Attenua, who is using an α7nAChR agonist, bradanicline, in Phase 2 clinical trials for chronic cough to electroceutical companies. The latter group of companies is very competitive, all of whom are developing devices to stimulate the vagus nerve as a therapy for inflammation. For example, Endonovo Therapeutics has developed a non-invasive electroceutical device using pulsed short-wave radiofrequency at 27.12 MHz that has been FDA-cleared and CE Marked for the palliative treatment of soft tissue injuries and post-operative pain and edema, and has Centers for Medicare and Medicaid Services (formerly the Health Care Financing Administration) (“CMS”) national coverage for the treatment of chronic wounds. Additionally, SetPoint Medical Corp is using vagal nerve stimulation for IBD and RA.
The electroceutical companies can be viewed as competition, or a vast proof-of-concept. Because in many respects, the α7nAChR program can be viewed as a chemical stimulation of the vagus nerve, and each of the indications benefiting from electrical stimulation, should be amenable to chemical stimulation.
Lastly, each of the large pharmaceutical companies that initially developed α7nAChR agonists could revitalize their programs and use their drugs in clinical trials for inflammatory indications.
Government Regulation
We have obtained regulatory approvals from the U.K. Medicines and Healthcare Products Regulatory Agency (MHRA) and the Dutch Centrale Commissie Mensgebonden Onderzoek (CCMO), as well as from the relevant accredited ethics committees, in order to perform clinical trials in the U.K. and The Netherlands solely for indications under the anti-TNF platform. We have not held any meetings with, and no applications or requests for approval have been submitted to, the U.S. Food and Drug Administration (“FDA”) for any indications or products under the anti-TNF platform at this time.
FDA Approval Process
In the U.S., pharmaceutical products, including drugs and biologics, are subject to extensive regulation by FDA. Under the U.S. Federal Food, Drug, and Cosmetic Act (the “FDC Act”), a “drug” is defined to include “articles intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease in man or other animals” and “articles (other than food) intended to affect the structure or any function of the body of man or other animals.” 21 USC 321(g). Like all drugs, biological products are also used for the treatment, prevention or cure of disease in humans. However, in contrast to chemically synthesized small molecular weight drugs, which have a well-defined structure and can be thoroughly characterized, biological products are generally derived from living material, such as human, animal, or microorganism, are complex in structure, and thus are always fully characterized. The U.S. Public Health Servicer Act (the PHS Act”) defines a biological product as a “virus, therapeutic serum, toxin, antitoxin, vaccine, blood, blood component or derivative, allergenic product, or analogous product … applicable to the prevention, treatment, or cure of a disease or condition of human beings.” 42 USC 262(i). FDA regulations and policies have established that biological products include blood-derived products, vaccines, in vivo diagnostic allergenic products, immunoglobulin products, products containing cells or microorganisms, and most protein products. Biological products subject to the PHS Act also meet the definition of drugs under the FDC Act. Biological products are a subset of drugs, and therefore both are regulated under provisions of the FDC Act. However, only biological products are licensed under section 351 of the PHS Act, although some therapeutic protein products have been approved under section 505 of the FDC Act rather than the PHS Act.)
26
The FDC Act, PHS Act, and other federal and state statutes and regulations, govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling, and import and export of drugs and biologics. Failure to comply with applicable U.S. requirements may subject a company to a variety of administrative or judicial sanctions, such as imposition of clinical holds, FDA refusal to approve pending NDAs/BLAs or supplements to approved NDAs/BLAs, withdrawal of approvals, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties and criminal prosecution.
Drug and biologic development in the U.S. typically involves pre-clinical laboratory and animal tests, the submission to the FDA of an IND, which must become effective before clinical testing may commence. For commercial approval, the sponsor must submit adequate tests by all methods reasonably applicable to show that the drug/biologic is safe for use under the conditions prescribed, recommended or suggested in the proposed labeling. The sponsor must also submit substantial evidence, generally consisting of adequate, well-controlled clinical trials to establish that the drug/biologic will have the effect it purports or is represented to have under the conditions of use prescribed, recommended or suggested in the proposed labeling, including meeting FDA standards for safety, and efficacy for drugs or purity and potency for biologics. Satisfaction of FDA pre-market approval requirements typically takes many years and the actual time required may vary substantially based upon the type, complexity and novelty of the product candidate or disease.
Pre-clinical tests include laboratory evaluation of product candidate chemistry, formulation and toxicity, as well as animal trials to assess the characteristics and potential safety and efficacy of the product candidate. The conduct of the pre-clinical tests must comply with federal regulations and requirements, including FDA’s GLP regulations and the U.S. Department of Agriculture’s regulations implementing the Animal Welfare Act of 1996. The results of pre-clinical testing are submitted to the FDA as part of an IND along with other information, including information about product candidate chemistry, manufacturing and controls, and a proposed clinical trial protocol. Long-term pre-clinical tests, such as animal tests of reproductive toxicity and carcinogenicity, may continue after the IND is submitted.
A 30-day waiting period after the submission of each IND is required prior to the commencement of clinical testing in humans. If the FDA has not imposed a clinical hold on the IND or otherwise commented or questioned the IND within this 30-day period, the IND is deemed issued, and the clinical trial proposed in the IND may begin.
Clinical trials involve the administration of the investigational new drug/biologic to healthy volunteers or patients under the supervision of a qualified investigator. Clinical trials must be conducted: (i) in compliance with GCP, an international standard and U.S. legal requirement meant to protect the rights and health of patients and to define the roles of clinical trial sponsors, administrators and monitors, (ii) in compliance with other federal regulations, and (iii) under protocols detailing the objectives of the trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. Each protocol involving testing on U.S. patients and subsequent protocol amendments must be submitted to the FDA as part of the IND.
27
The FDA may order the temporary, or permanent, discontinuation of a clinical trial at any time or impose other sanctions if it believes that the clinical trial either is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial patients. The trial protocol and informed consent information for patients in clinical trials must also be submitted to an Institutional Review Board (“IRB”), for approval. An IRB may also prevent a clinical trial from beginning or require the clinical trial at the site to be halted, either temporarily or permanently, for failure to comply with the IRB’s requirements or may impose other conditions.
Clinical trials to support NDAs/BLAs for marketing approval are typically conducted in three sequential phases, but the phases may overlap or otherwise vary in particular circumstances. In Phase 1, the initial introduction of the drug/biologic into healthy human subjects or patients, the drug/biologic is tested to assess metabolism, pharmacokinetics, pharmacological actions, side effects associated with increasing doses and, if possible, early evidence on effectiveness. Phase 2 usually involves trials in a limited patient population to determine the effectiveness of the drug/biologic for a particular indication, dosage tolerance and optimum dosage, and to identify common adverse effects and safety risks. If a compound demonstrates evidence of effectiveness and an acceptable safety profile in Phase 2 evaluations, Phase 3 trials are undertaken to obtain the additional information about clinical efficacy and safety in a larger number of patients, typically at geographically dispersed clinical trial sites, to permit the FDA to evaluate the overall benefit-risk relationship of the drug/biologic and to provide adequate information for the labeling of the drug/biologic. In most cases, the FDA requires two adequate and well-controlled Phase 3 clinical trials to demonstrate the efficacy of the drug/biologic. The FDA may, however, determine that a single Phase 3 trial with other confirmatory evidence may be sufficient in some instances. In some cases, the FDA may require post-market studies, known as Phase 4 studies, to be conducted as a condition of approval in order to gather additional information on the drug’s/biologic’s effect in various populations and any side effects associated with long-term use. Depending on the risks posed by the drugs/biologics, other post-market requirements may be imposed.
After completion of the required clinical testing, a New Drug Application (“NDA”)/Biologics License Application (“BLA”) is prepared and submitted to the FDA. The FDA approval of the NDA/BLA is required before marketing of the product candidate may begin in the U.S. The NDA/BLA must include the results of all pre-clinical, clinical, and other testing and a compilation of data relating to the product candidate’s pharmacology, chemistry, manufacture, and controls. The cost of preparing and submitting an NDA/BLA is substantial. Under federal law, the submission of most NDAs/BLAs is also subject to an application user fee, which, for the fiscal year 2022, was in the amount of approximately $3.1 million.
The FDA has 60 days from its receipt of an NDA/BLA to determine whether the application will be accepted for filing based on the agency’s threshold determination that it is sufficiently complete to permit substantive review. Once the submission is accepted for filing, the FDA begins an in-depth review. Under the Prescription Drug User Fee Act, the FDA has agreed to certain performance goals in the review of NDAs/BLAs. The FDA’s current performance goals call for the FDA to complete review of 90 percent of standard (non-priority) NDAs/BLAs within 10 months of receipt and within six months for priority NDAs/BLAs, but two additional months are added to standard and priority NDAs/BLAs for a new molecular entity/reference biologic. A drug/biologic is eligible for priority review if it addresses an unmet medical need in a serious or life-threatening disease or condition. The review process for both standard and priority review may be extended by FDA for three additional months to consider certain late-submitted information, or information intended to clarify information already provided in the submission. These timelines are not legally binding on the FDA.
The FDA may also refer applications for novel drug/biologic products, or drug/biologic products that present difficult questions of safety or efficacy, to an advisory committee, which is typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations. Before approving an NDA/BLA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP.
28
Additionally, the FDA will inspect the facility or the facilities at which the drug is manufactured. The FDA will not approve the product candidate unless compliance with Good Manufacturing Practice regulations (“GMPs”), is satisfactory and the NDA/BLA contains data that provide substantial evidence that the drug is safe and effective, or the biologic meets the standards for safety, purity, and potency in the indication studied.
After the FDA evaluates the NDA/BLA and the manufacturing facilities, the FDA issues either an approval letter or a complete response letter. A complete response letter generally outlines the deficiencies in the submission and may require substantial additional testing, or information, in order for the FDA to reconsider the application. If, or when, those deficiencies have been addressed to the FDA’s satisfaction in a resubmission of the NDA/BLA, the FDA will issue an approval letter. The FDA has committed to reviewing 90 percent of resubmissions within two or six months depending on the type of information included. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
An approval letter authorizes commercial marketing of the drug/biologic with specific prescribing information for specific indications. As a condition of NDA/BLA approval, the FDA may require a Risk Evaluation and Mitigation Strategy (“REMS”), to help ensure that the benefits of the drug/biologic outweigh the potential risks. REMS can include medication guides, communication plans for health care professionals, and Elements to Assure Safe Use (ETASU). ETASU can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring, and the use of patient registries. The requirement for a REMS can materially affect the potential market and profitability of the drug/biologic. Moreover, product candidate approval may require substantial post approval testing and surveillance to monitor the drug’s/biologic’s safety or efficacy. Once granted, product candidate approvals may be withdrawn if compliance with regulatory standards is not maintained or problems are identified following initial marketing.
Disclosure of Clinical Trial Information
Sponsors of clinical trials of certain FDA-regulated products, including prescription drugs/biologics, are required to register and disclose certain clinical trial information on a public website maintained by the U.S. National Institutes of Health. Information related to the product candidate, patient population, phase of investigation, study sites and investigator, and other aspects of the clinical trial is made public as part of the registration. Sponsors are also obligated to disclose the results of these trials after completion. The deadline for submitting the results of these trials can be extended for up to two years if the sponsor certifies that it is seeking approval of an unapproved product or that it will file an application for approval of a new indication for an approved product within one year. Competitors may use the publicly available information to gain knowledge regarding the design and progress of our development programs.
Fast Track Designation and Accelerated Approval
If our drug/biologic candidate meets the requirements of the FDA’s fast track program, we would seek to have our drug/biologic candidate expedited through this program. The FDA has programs to facilitate the development, and expedite the review, of drugs/biologics that are intended for the treatment of a serious or life-threatening disease or condition for which there is no effective treatment and which demonstrate the potential to address unmet medical needs for the condition. Under the fast-track program, the sponsor of a new drug/biologic candidate may request that the FDA designate the drug/biologic candidate for a specific indication as a fast track drug/biologic concurrent with, or after, the filing of the IND for the drug/biologic candidate. The FDA must determine if the drug/biologic candidate qualifies for fast track designation within 60 days of receipt of the sponsor’s request. In addition to other benefits such as the ability to engage in more frequent interactions with the FDA, the FDA may initiate review of sections of a fast track drug’s/biologic’s NDA/BLA before the application is complete. This rolling review is available if the applicant provides, and the FDA approves, a schedule for the submission of the remaining information and the applicant pays applicable user fees. However, the FDA’s time period goal for reviewing an application does not begin until the last section of the NDA/BLA is submitted. Additionally, the fast track designation may be withdrawn by the FDA if it believes that the designation is no longer supported by data emerging in the clinical trial process.
29
Under the FDA’s accelerated approval regulations, the FDA may approve a drug/biologic for a serious or life-threatening illness that provides meaningful therapeutic benefit to patients over existing treatments based upon a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity or prevalence of the condition and the availability or lack of alternative treatments.
In clinical trials, a surrogate endpoint is a measurement of laboratory or clinical signs of a disease or condition that substitutes for a direct measurement of how a patient feels, functions or survives. Surrogate endpoints can often be measured more easily or more rapidly than clinical endpoints. A drug/biologic candidate approved on this basis is subject to rigorous post-marketing compliance requirements, including the completion of Phase 4 or post- approval clinical trials to confirm clinical benefit. Failure to conduct required post-approval studies, or confirm a clinical benefit during post-marketing studies, will allow the FDA to withdraw the drug/biologic from the market on an expedited basis. Unless otherwise informed by the FDA, for an accelerated approval product/biologic, an applicant must submit to the FDA for consideration during the preapproval review period copies of all promotional materials, including promotional labeling as well as advertisements, intended for dissemination or publication within 120 days following marketing approval. After 120 days following marketing approval, unless otherwise informed by the FDA, the applicant must submit promotional materials at least 30 days prior to the intended time of initial dissemination of the labeling or initial publication of the advertisement.
Breakthrough Therapy Designation
As with the FDA’s fast track program, if our drug/biologic candidate meets the requirements to receive the FDA’s Breakthrough Therapy designation, we would seek to have our drug/biologic candidate expedited through this program. The FDA’s Breakthrough Therapy designation program is intended to expedite the development and review of products that treat serious or life-threatening diseases or conditions. A Breakthrough Therapy is defined, under the Food and Drug Administration Safety and Innovation Act, as a drug/biologic that is intended, alone or in combination with one or more other drugs/biologics, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the drug/biologic may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. The designation includes all of the features of fast-track designation, as well as more intensive FDA interaction and guidance. The Breakthrough Therapy designation is a distinct status from both accelerated approval and priority review, but these can also be granted to the same product candidate if the relevant criteria are met. The FDA must take certain actions, such as holding timely meetings and providing advice, intended to expedite the development and review of an application for approval of a breakthrough therapy. All requests for breakthrough therapy designation will be reviewed within 60 days of receipt, and the FDA will either grant or deny the request.
Fast track designation, accelerated approval, priority review, and Breakthrough Therapy designation do not change the standards for approval but may expedite the development or approval process. Even if the FDA grants one of these designations, the FDA may later decide that the drug/biologic products no longer meet the conditions for qualification.
Orange Book Listing and Patent Certification
Based on amendments to the FDC Act made by the Drug Price Competition and Innovation Act of 1984 (commonly known as Hatch-Waxman), in seeking approval for a drug through an NDA, applicants are required to list with the FDA each patent whose claims cover the applicant’s product candidate or a claimed method of use of the product candidate. Upon approval of a drug, each of the eligible patents listed in the application for the drug is then published in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book. Drugs listed in the Orange Book must, in turn, be the subject of a special certification by the filer of an abbreviated new drug application (“ANDA”), for a generic version of the drug, or by the applicant of a hybrid application known as a 505(b)(2) application. An ANDA provides for marketing of a drug product candidate that has the same active ingredient(s) in the same strengths and dosage form as the reference listed innovator drug and has been shown to be bioequivalent to the reference listed drug. Other than the requirement for bioequivalence testing (absent a waiver), ANDA applicants are not required to conduct, or submit results of, pre-clinical or clinical tests to prove the safety or effectiveness of their drug product candidate. Drugs approved in this way are commonly referred to as “generic equivalents” to the listed drug, are considered therapeutically equivalent to the listed drug, and can often be substituted by pharmacists under prescriptions written for the original listed drug.
30
The ANDA applicant is required to certify to the FDA concerning any patents listed for the approved product candidate in the FDA’s Orange Book. Specifically, the applicant must certify that: (i) the required patent information has not been filed; (ii) the listed patent has expired; (iii) the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or (iv) the listed patent is invalid or will not be infringed by the new product candidate. The ANDA applicant may also elect to submit a “section viii statement”, certifying that its proposed ANDA labeling does not contain (or carves out) any language regarding the patented method-of- use, rather than certify to a listed method-of-use patent.
If the applicant does not challenge the listed patents, the ANDA application will not be approved until all the listed patents claiming the referenced product have expired.
A certification that the new product candidate will not infringe the already approved product candidate’s listed patents, or that such patents are invalid or unenforceable, is called a Paragraph IV certification. If the ANDA applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the NDA and patent holders once the ANDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the Paragraph IV certification. The filing of a patent infringement lawsuit within 45 days of the receipt of notice of a Paragraph IV certification automatically prevents the FDA from approving the ANDA until the earlier of 30 months, expiration of the patent, settlement of the lawsuit, a decision in the infringement case that is favorable to the ANDA applicant, or some other order of the court.
Sponsors may also seek to market versions of drug products via a section 505(b)(2) application, which is an NDA pathway that allows an applicant to seek approval for a drug product based on full safety and efficacy documentation, some of which may be from literature or conducted by others and for which the applicant does not have the right of reference. NDA Section 505(b)(2) applications may be submitted for drug products that represent a modification, such as a new indication or new dosage form, of a previously approved drug. Section 505(b)(2) applications may rely on the FDA’s previous findings for the safety and effectiveness of the previously approved drug in addition to information obtained by the 505(b)(2) applicant to support the modification of the previously approved drug. Preparing Section 505(b)(2) applications may be less costly and time-consuming than preparing an NDA based entirely on new data and information. Section 505(b)(2) applications are subject to the same patent certification procedures as an ANDA.
New Chemical Entity Exclusivity and Clinical Investigation Exclusivity
Upon NDA approval of a new chemical entity (“NCE”), which is a drug that contains no active moiety that has been approved by the FDA in any other NDA, that drug receives five years of marketing exclusivity during which time the FDA cannot receive any ANDA or 505(b)(2) application seeking approval of a drug that references a version of the NCE drug. Certain changes to a drug, such as the addition of a new indication to the package insert with new clinical studies required for approval, are associated with a three-year period of exclusivity during which the FDA cannot approve an ANDA or 505(b)(2) application that includes the change.
An ANDA or 505(b)(2) application may be submitted one year before NCE exclusivity expires if a Paragraph IV certification is filed. If there is no listed patent in the Orange Book, there may not be a Paragraph IV certification and thus no ANDA or 505(b)(2) may be filed before the expiration of the exclusivity period.
31
For a botanical drug, FDA may determine that the active moiety is one or more of the principle components or the complex mixture as a whole. This determination would affect the utility of any five-year exclusivity as well as the ability of any potential generic competitor to demonstrate that it is the same drug as the original botanical drug.
Five-year and three-year exclusivities do not preclude FDA approval of a 505(b)(1) application for a version of the drug during the period of exclusivity, provided that the 505(b)(1) conducts or obtains a right of reference to all of the pre-clinical studies and adequate and well controlled clinical trials necessary to demonstrate safety and effectiveness.
Orphan Designation and Exclusivity
FDA may grant orphan drug designation to drugs intended to treat a rare disease or condition that affects fewer than 200,000 individuals in the U.S., or if it affects more than 200,000 individuals in the U.S. and there is no reasonable expectation that the cost of developing and making the drug for this type of disease or condition will be recovered from sales in the U.S.
Orphan drug designation entitles a party to financial incentives such as opportunities for grant funding towards clinical study costs, tax advantages, and user-fee waivers. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. In addition, the first NDA or BLA applicant to receive orphan drug designation for a particular drug is entitled to orphan drug exclusivity, which means the FDA may not approve any other application to market the same drug for the same indication for a period of seven years in the U.S., except in limited circumstances. Orphan drug exclusivity does not prevent the FDA from approving a different drug for the same disease or condition, or the same drug for a different disease or condition.
Pediatric Studies and Exclusivity
NDAs and BLAs must contain data to assess the safety and effectiveness of an investigational new drug product for the claimed indications in all relevant pediatric populations in order to support dosing and administration for each pediatric subpopulation for which the drug is safe and effective. The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults or full or partial waivers if certain criteria are met. Discussions about pediatric development plans can be discussed with the FDA at any time, but usually occur any time between the end-of-Phase 2 meeting and submission of the NDA or BLA. Unless otherwise required by regulation, the requirements for pediatric data do not apply to any drug for an indication for which orphan designation has been granted.
Pediatric exclusivity is another type of non-patent exclusivity in the U.S. that may be granted if certain FDA requirements are met, such as FDA’s determination that information relating to the use of a new drug in the pediatric population may produce health benefits, and the applicant agrees to perform and report on FDA-requested studies within a certain time frame. Pediatric exclusivity adds a period of six months of exclusivity to the end of all existing marketing exclusivity and patents held by the sponsor for that active moiety. This is not a patent term extension, but it effectively extends the regulatory period during which the FDA cannot accept or approve another application relying on the NDA or BLA sponsor’s data.
Biologics Exclusivity and Biosimilars
The Patient Protection and Affordable Care Act of 2010, or Affordable Care Act, includes a subtitle called the Biologics Price Competition and Innovation Act of 2009, or BPCI Act, which amended the PHS Act to create an abbreviated approval pathway under section 351(k) of the PHS Act for biological products shown to be similar to, or interchangeable with, an FDA-licensed reference biological product originally licensed under section 351(a) of the PHS Act.
32
A reference biologic is granted twelve years of marketing exclusivity from the time of first licensure of the reference product, during which time a 351(k) application for a biosimilar of the reference product may not be approved. The reference biologic is also granted four years of so-called data exclusivity, during which time a 351(k) application for a biosimilar of the reference product may not be submitted for review. The first biologic product submitted under the abbreviated approval pathway that is determined to be interchangeable with the reference product has exclusivity against other biologics submitting under the abbreviated approval pathway for the lesser of (i) one year after the first commercial marketing, (ii) eighteen months after approval if there is no legal challenge, (iii) eighteen months after the resolution in the applicant’s favor of a lawsuit challenging the biologics’ patents if an application has been submitted, or (iv) 42 months after the application has been approved if a lawsuit is ongoing within the 42-month period.
Biologic Patent Information
In contrast to small molecule drugs, for which applicants are required to submit patent information with their NDAs and certain supplements, an applicant seeking licensure of a biological product need not submit patent information in its BLA or supplements. Also, unlike small molecule drugs, for which the approvability of ANDAs and 505(b)(2) NDAs is impacted by the status of listed patents for the reference NDA drug product, the approvability of section 351(k) applications for biosimilar products is presently delinked from the various processes for resolving patent disputes. Biosimilar applicants have a choice whether to engage in the patent litigation provisions of the BPCIA, colloquially known as the “patent dance,” to identify and litigate a defined list of patents. However, unlike the listing of small molecule reference listed drugs and patents in the “Orange Book,” there had not been a process for listing patents in FDA’s List of Licensed Biological Products, commonly known as the “Purple Book.” Recently, in December 2020 Congress enacted the Biological Product Patent Transparency Act (“BPPT”) (originally introduced as the Purple Book Continuity Act created section 351(k)(9) of the PHS Act. That section requires that a biological product reference sponsor that provides a biosimilar applicant with a patent list as part of the “Patent Dance” BPCIA patent litigation process must now submit those lists to FDA within 30 days, and further, as of June 2021 FDA is required to make those lists (along with any revisions or updates) public in the Purple Book database.
Patent Term Extension
After NDA or BLA approval, owners of relevant drug or biologic patents may apply for up to a five-year patent extension. The allowable patent term extension is calculated as half of the product’s testing phase — the time between IND submission and NDA or BLA submission — and all of the review phase — the time between NDA submission and approval up to a maximum of five years. The time can be shortened if FDA determines that the applicant did not pursue approval with due diligence. The total patent term after the extension may not exceed 14 years.
For patents that might expire during the application phase, the patent owner may request an interim patent extension. An interim patent extension increases the patent term by one year and may be renewed up to four times. For each interim patent extension granted, the post-approval patent extension is reduced by one year. The director of the United States Patent and Trademark Office (USPTO) must determine that approval of the product covered by the patent for which a patent extension is being sought is likely. Interim patent extensions are not available for a drug for which an NDA or BLA has not been submitted.
Advertising and Promotion
Once an NDA or BLA is approved, a product candidate will be subject to certain post-approval requirements. For instance, FDA closely regulates the post-approval marketing and promotion of drugs and biologics, including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities and promotional activities involving the internet.
Drugs and biologics may be marketed only for the approved indications and in accordance with the provisions of the approved labeling.
33
Post-Approval Changes
Changes to some of the conditions established in an approved application, including changes in indications, labeling, or manufacturing processes or facilities, require submission and FDA approval of a new NDA/BLA or NDA/BLA supplement before the change can be implemented. An NDA supplement for a new indication typically requires clinical data similar to that in the original application, and the FDA uses the same procedures and actions in reviewing NDA supplements as it does in reviewing NDAs.
Adverse Event Reporting and GMP Compliance
Adverse event reporting on an expedited basis and submission of periodic adverse event reports is required following FDA approval of an NDA or BLA. The FDA also may require post-marketing testing, known as Phase 4 testing, REMS and surveillance to monitor the effects of an approved product, or the FDA may place conditions on an approval that could restrict the distribution or use of the product. In addition, quality-control, drug manufacture, packaging, and labeling procedures must continue to conform GMPs after approval. Drug manufacturers and certain of their subcontractors are required to register their establishments with FDA and certain state agencies. Registration with the FDA subjects entities to periodic unannounced inspections by the FDA, during which the agency inspects manufacturing facilities to assess compliance with GMPs. Accordingly, manufacturers must continue to expend time, money and effort in the areas of production and quality control to maintain compliance with GMPs. Regulatory authorities may withdraw product approvals, issue warning letters, request product recalls or take other enforcement actions if a company fails to comply with regulatory standards, if it encounters problems following initial marketing or if previously unrecognized problems are subsequently discovered.
Special Protocol Assessment
A sponsor may reach an agreement with the FDA under the Special Protocol Assessment (“SPA”), process as to the required design and size of clinical trials intended to form the primary basis of an efficacy claim. According to its performance goals, the FDA has committed to evaluating 90 percent of the protocols within 45 days of its receipt of the requests to assess whether the proposed trial is adequate, and that evaluation may result in discussions and a request for additional information. An SPA request must be made before the proposed trial begins, and all open issues must be resolved before the trial begins. If a written agreement is reached, it will be documented and made part of the administrative record. Under the FDC Act and FDA guidance implementing the statutory requirement, an SPA is generally binding upon the FDA as to the design of the trial except in limited circumstances, such as if the FDA identifies a substantial scientific issue essential to determining safety or efficacy after the study begins, public health concerns emerge that were unrecognized at the time of the protocol assessment, the sponsor and FDA agree to the change in writing, or if the study sponsor fails to follow the protocol that was agreed upon with the FDA.
Controlled Substances
The CSA and the implementing regulations impose registration, security, recordkeeping and reporting, storage, manufacturing, distribution, dispensing, importation and other requirements on controlled substances under the oversight of the U.S. Drug Enforcement Administration (“DEA”). The DEA is the federal agency, responsible for regulating controlled substances, and requires those individuals or entities that manufacture, import, export, distribute, research, or dispense controlled substances to comply with the regulatory requirements in order to prevent the diversion and abuse of controlled substances.
The DEA regulates controlled substances as Schedule I, II, III, IV or V substances, depending on the substance’s medical effectiveness and abuse potential. Pharmaceutical products having a currently accepted medical use that are otherwise approved for marketing may be listed as Schedule II, III, IV or V substances, with Schedule II substances presenting the highest potential for abuse and physical or psychological dependence, and Schedule V substances presenting the lowest relative potential for abuse and dependence. The DEA has placed certain drug products that include cannabidiol, on Schedule V.
34
Following NDA approval of a drug containing a Schedule I controlled substance, that substance must be rescheduled as a Schedule II, III, IV or V substance before it can be marketed. The Improving Regulatory Transparency for New Medical Therapies Act enacted on November 25, 2015 and its implementing regulations has removed uncertainty associated with the timing of the DEA rescheduling process after NDA approval, under which a manufacturer may market its product no later than 90 days after the later of: (1) the date on which DEA receives from FDA the scientific and medical evaluation and scheduling recommendation; or (2) the date on which DEA receives from FDA notification that FDA has approved the drug. The Act also clarifies that the seven-year orphan exclusivity period begins with the approval of the NDA or DEA scheduling, whichever is later. This changes the previous situation whereby the orphan “clock” began to tick upon FDA’s NDA approval, even though the product could not be marketed until DEA scheduling was complete.
The CSA requires that facilities that manufacture, distribute, dispense, import or export any controlled substance must register annually with the DEA. Separate registrations are required for importation and manufacturing activities, and each registration authorizes the specific schedules of controlled substances the registrant may handle. Prior to issuance of a controlled substance registration, the DEA inspects all manufacturing facilities to review security, recordkeeping, reporting and handling of the controlled substances. The specific security requirements vary by, among other things, the type of business activity conducted, and the type, form, and quantity of controlled substances handled.
In addition, the states have their own distinct controlled substance laws and regulations, including licensure, distribution, dispensing, recordkeeping and reporting requirements for controlled substances. State boards of pharmacy or similar authorities regulate use of controlled substances in each state. Failure to comply with applicable requirements, such as the loss or diversion of controlled substances, can result in administrative fines, suspension or revocation of licenses, and civil and criminal liabilities.
U.K./Europe/Rest of World Government Regulation
In addition to regulations in the U.S., we are and will be subject, either directly or through our distribution partners, to a variety of regulations in other jurisdictions governing, among other things, clinical trials and any commercial sales (including pricing and reimbursement) and distribution of our products, if approved.
Whether or not we obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in non-U.S. countries prior to the commencement of clinical trials or marketing of the product in those countries.
In the EU, medicinal products are subject to extensive pre- and post-marketing regulation by regulatory authorities at both the EU and national levels. Additional rules also apply at the national level to the manufacture, import, export, storage, distribution and sale of controlled substances. In many EU member states the regulatory authority responsible for medicinal products is also responsible for controlled substances. Responsibility is, however, split in some member states. Generally, any company manufacturing or distributing a medicinal product containing a controlled substance in the EU will need to hold a controlled substances license from the competent national authority and will be subject to specific record-keeping and security obligations. Separate import or export certificates are required for each shipment into or out of the member state.
In the U.K., medicinal products are subject to extensive regulation by the Medicines and Healthcare products Regulatory Agency (“MHRA”), which is an executive agency, sponsored by the Department of Health and Social Care. MHRA regulates by ensuring that medicines, medical devices and blood components for transfusion meet applicable standards of safety, quality and efficacy, in addition to supporting innovation and research and development that is beneficial to public health.
35
Clinical Trials and Marketing Approval
Certain countries outside of the U.S. have a process that requires the submission of a clinical trial application much like an IND prior to the commencement of human clinical trials. In Europe, for example, a clinical trial application (“CTA”), must be submitted to the competent national health authority and to independent ethics committees in each country in which a company intends to conduct clinical trials. Once the CTA is approved in accordance with a country’s requirements and a company has received favorable ethics committee approval, clinical trial development may proceed in that country.
Requirements for the conduct of clinical trials in the U.K. and EU, including GCP, are implemented in the Clinical Trials Directive 2001/20/EC and the GCP Directive 2005/28/EC. Pursuant to Directive 2001/20/EC and Directive 2005/28/EC, as amended, a system for the approval of clinical trials in the EU has been implemented through national legislation of EU member states. Under this system, approval must be obtained from the relevant competent national authority of each EU member state in which a clinical trial is planned. A CTA must be submitted and supported by an investigational medicinal product dossier along with additional supporting information pursuant to Directive 2001/20/EC, Directive 2005/28/EC and other applicable guidance documents. Furthermore, a clinical trial may only commence after a competent ethics committee has issued a favorable opinion on the clinical trial application in the U.K. or the specific EU member state.
In April 2014, the Clinical Trials Regulation, Reg. (EU) No 536/2014 (the “New Regulation”) was adopted to replace the Clinical Trials Directive 2001/20/EC (the “Prior Directive”). To ensure that the rules for clinical trials are identical throughout the EU, new EU clinical trials legislation was passed as a “regulation” that is directly applicable to EU member states. A new, single CTA is planned for all EU member states, which will be submitted via an online portal to streamline the authorization process. Until the New Regulation is fully implemented, clinical trials will continue to be implemented under the Prior Directive.
The requirements and process governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country, even though there is already some degree of legal harmonization in the EU member states resulting from the national implementation of underlying EU legislation. In all cases, the clinical trials must be conducted in accordance with the International Conference on Harmonization guidelines on GCP and other applicable regulatory requirements.
To obtain regulatory approval to place a drug on the market in the U.K. or EU countries, we must submit a marketing authorization application. This application is similar to the NDA in the U.S., with the exception of, among other things, country-specific document requirements. All application procedures require an application in the common technical document, format, which includes the submission of detailed information about the manufacturing and quality of the product, and nonclinical and clinical trial information. Drugs can be authorized in the U.K. or EU by using (i) the centralized authorization procedure, (ii) the mutual recognition procedure, (iii) the decentralized procedure or (iv) national authorization procedures.
The European Commission created the centralized procedure for the approval of human drugs to facilitate marketing authorizations that are valid throughout the U.K. and EU and, by extension (after national implementing decisions) in Iceland, Liechtenstein and Norway, which, together with the EU member states, comprise the European Economic Area. Applicants file marketing authorization applications with the European Medicines Agency (EMA), where they are reviewed by a relevant scientific committee, in most cases the Committee for Medicinal Products for Human Use (“CHMP”). The European Medicines Agency (“EMA”) forwards CHMP opinions to the European Commission, which uses them as the basis for deciding whether to grant a marketing authorization. This procedure results in a single marketing authorization granted by the European Commission that is valid across the EU, as well as in Iceland, Liechtenstein and Norway. The centralized procedure is compulsory for human drugs that are: (i) derived from biotechnology processes, such as genetic engineering, (ii) contain a new active substance indicated for the treatment of certain diseases, such as HIV/AIDS, cancer, diabetes, neurodegenerative diseases, autoimmune and other immune dysfunctions and viral diseases, (iii) officially designated “orphan drugs” (drugs used for rare human diseases) and (iv) advanced-therapy medicines, such as gene-therapy, somatic cell-therapy or tissue-engineered medicines. The centralized procedure may at the voluntary request of the applicant also be used for human drugs that do not fall within the above-mentioned categories if the CHMP agrees that the human drug (a) contains a new active substance not yet approved on November 20, 2005; (b) constitutes a significant therapeutic, scientific or technical innovation or (c) authorization under the centralized procedure is in the interests of patients at the EU level.
36
Under the centralized procedure in the EU, the maximum time frame for the evaluation of a marketing authorization application by the EMA is 210 days (excluding clock stops, when additional written or oral information is to be provided by the applicant in response to questions asked by the CHMP), with adoption of the actual marketing authorization by the European Commission thereafter.
Accelerated evaluation might be granted by the CHMP in exceptional cases, when a medicinal product is expected to be of a major public health interest from the point of view of therapeutic innovation, defined by three cumulative criteria: the seriousness of the disease to be treated; the absence of an appropriate alternative therapeutic approach, and anticipation of exceptional high therapeutic benefit. In this circumstance, EMA ensures that the evaluation for the opinion of the CHMP is completed within 150 days and the opinion issued thereafter.
For those medicinal products for which the centralized procedure is not available, the applicant must submit marketing authorization applications to the national medicines regulators through one of three procedures: (i) the mutual recognition procedure (which must be used if the product has already been authorized in at least one other EU member state, and in which the EU member states are required to grant an authorization recognizing the existing authorization in the other EU member state, unless they identify a serious risk to public health), (ii) the decentralized procedure (in which applications are submitted simultaneously in two or more EU member states) or (iii) national authorization procedures (which results in a marketing authorization in a single EU member state).
Mutual Recognition Procedure
The mutual recognition procedure (“MRP”), for the approval of human drugs is an alternative approach to facilitate individual national marketing authorizations within the U.K. and EU. Basically, the MRP may be applied for all human drugs for which the centralized procedure is not obligatory. The MRP is applicable to the majority of conventional medicinal products, and must be used if the product has already been authorized in one or more member states.
The characteristic of the MRP is that the procedure builds on an already-existing marketing authorization in the U.K. or a member state of the EU that is used as a reference in order to obtain marketing authorizations in other EU member states. In the MRP, a marketing authorization for a drug already exists in one or more member states of the EU and subsequently marketing authorization applications are made in other EU member states by referring to the initial marketing authorization. The member state in which the marketing authorization was first granted will then act as the reference member state. The member states where the marketing authorization is subsequently applied for act as concerned member states. The concerned member states are required to grant an authorization recognizing the existing authorization in the reference member state, unless they identify a serious risk to public health.
The MRP is based on the principle of the mutual recognition by EU member states of their respective national marketing authorizations. Based on a marketing authorization in the reference member state, the applicant may apply for marketing authorizations in other member states. In such case, the reference member state shall update its existing assessment report about the drug in 90 days. After the assessment is completed, copies of the report are sent to all member states, together with the approved summary of product characteristics, labeling and package leaflet. The concerned member states then have 90 days to recognize the decision of the reference member state and the summary of product characteristics, labeling and package leaflet. National marketing authorizations shall be granted within 30 days after acknowledgement of the agreement.
37
Should any member state refuse to recognize the marketing authorization by the reference member state, on the grounds of potential serious risk to public health, the issue will be referred to a coordination group. Within a timeframe of 60 days, member states shall, within the coordination group, make all efforts to reach a consensus. If this fails, the procedure is submitted to an EMA scientific committee for arbitration. The opinion of this EMA Committee is then forwarded to the European Commission, for the start of the decision-making process. As in the centralized procedure, this process entails consulting various European Commission Directorates General and the Standing Committee on Human Medicinal Products.
Data Exclusivity
In the U.K. and EU, marketing authorization applications for generic medicinal products do not need to include the results of pre-clinical and clinical trials, but instead can refer to the data included in the marketing authorization of a reference product for which regulatory data exclusivity has expired. If a marketing authorization is granted for a medicinal product containing a new active substance, that product benefits from eight years of data exclusivity, during which generic marketing authorization applications referring to the data of that product may not be accepted by the regulatory authorities, and a further two years of market exclusivity, during which such generic products may not be placed on the market. The two-year period may be extended to three years if during the first eight years a new therapeutic indication with significant clinical benefit over existing therapies is approved.
Orphan Medicinal Products
The EMA’s Committee for Orphan Medicinal Products (“COMP”), may recommend orphan medicinal product designation to promote the development of products that are intended for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions affecting not more than five in 10,000 persons in the EU. Additionally, orphan designation is granted for products intended for the diagnosis, prevention or treatment of a life-threatening, seriously debilitating or serious and chronic condition and when, without incentives, it is unlikely that sales of the product in the U.K. and EU would be sufficient to justify the necessary investment in developing the medicinal product. The COMP may only recommend orphan medicinal product designation when the product in question offers a significant clinical benefit over existing approved products for the relevant indication. Following a positive opinion by the COMP, the European Commission generally grants orphan status within 30 days. When the draft decision of the European Commission is not aligned with the COMP opinion, the COMP will reassess orphan status in parallel with EMA review of a marketing authorization application and orphan status may be withdrawn at if the drug no longer fulfills the orphan criteria (for instance, because a new product was approved for the indication and no data is available to demonstrate a significant benefit over that new product). Orphan medicinal product designation entitles a party to financial incentives such as reduction of fees or fee waivers and ten years of market exclusivity is granted following marketing authorization. During this period, the competent authorities may not accept or approve any similar medicinal product for the same therapeutic indication, unless it offers a significant clinical benefit or if the holder of the marketing authorization for the original orphan drug is unable to supply sufficient quantities of the drug. This period may be reduced to six years if the orphan medicinal product designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity.
Pediatric Development
In the EU and the U.K., companies developing a new medicinal product must agree to a Pediatric Investigation Plan (“PIP”), with the EMA or the MHRA and must conduct pediatric clinical trials in accordance with that PIP unless a waiver applies, for example, because the relevant disease or condition occurs only in adults. The marketing authorization application for the product must include the results of pediatric clinical trials conducted in accordance with the PIP, unless a waiver applies, or a deferral has been granted, in which case the pediatric clinical trials must be completed at a later date. Products that are granted a marketing authorization on the basis of the pediatric clinical trials conducted in accordance with the PIP are eligible for a six-month extension of the protection under a supplementary protection certificate (if the product covered by it qualifies for one at the time of approval). This pediatric reward is subject to specific conditions and is not automatically available when data in compliance with the PIP are developed and submitted.
38
If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension of clinical trials, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
In addition, most countries are parties to the Single Convention on Narcotic Drugs 1961 and the Convention on Psychotropic Substances 1971, which governs international trade and domestic control of narcotic substances, including cannabis extracts. Countries may interpret and implement their treaty obligations in a way that creates a legal obstacle to our obtaining marketing approval for our product candidates in those countries. These countries may not be willing or able to amend or otherwise modify their laws and regulations to permit our product candidates to be marketed, or achieving such amendments to the laws and regulations may take a prolonged period of time. In that case, we would be unable to market our products in those countries in the near future or perhaps at all.
In the U.K., medicinal products are subject to extensive regulation by the Medicines and Healthcare products Regulatory Agency (“MHRA”), which is an executive agency, sponsored by the Department of Health and Social Care. MHRA regulates by ensuring that medicines, medical devices and blood components for transfusion meet applicable standards of safety, quality and efficacy, in addition to supporting innovation and research and development that is beneficial to public health.
Reimbursement
Sales of pharmaceutical products in the U.S. will depend, in part, on the extent to which the costs of the products will be covered by third-party payers, such as government health programs, and commercial insurance and managed health care organizations. These third-party payers are increasingly challenging the prices charged for medical products and services. Additionally, the containment of health care costs has become a priority of federal and state governments, and the prices of drugs have been a focus in this effort. The U.S. government, state legislatures and foreign governments have shown significant interest in implementing cost-containment programs, including price controls, utilization management and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our net revenue and results. If these third-party payers do not consider our products to be cost effective compared to other available therapies, they may not cover our products after approval as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell our products on a profitable basis.
The Medicare Prescription Drug, Improvement, and Modernization Act of 2003 (“MMA”), imposed requirements for the distribution and pricing of prescription drugs for Medicare beneficiaries and included a major expansion of the prescription drug benefit under Medicare Part D. Under Part D, Medicare beneficiaries may enroll in prescription drug plans offered by private entities that provide coverage of outpatient prescription drugs. Part D is available through both stand-alone prescription drug benefit plans and prescription drug coverage as a supplement to Medicare Advantage plans. Unlike Medicare Parts A and B, Part D coverage is not standardized. Part D prescription drug plan sponsors are not required to pay for all covered Part D drugs, and each drug plan can develop its own drug formulary that identifies which drugs it will cover and at what tier or level. However, Part D prescription drug formularies must include drugs within each therapeutic category and class of covered Part D drugs, though not necessarily all the drugs in each category or class. Any formulary used by a Part D prescription drug plan must be developed and reviewed by a pharmacy and therapeutic committee. Government payment for some of the costs of prescription drugs may increase demand for products for which we receive marketing approval. However, any negotiated prices for our products covered by a Part D prescription drug plan will likely be lower than the prices we might otherwise obtain. Moreover, while the MMA applies only to drug benefits for Medicare beneficiaries, private payers often follow Medicare coverage policy and payment limitations in setting their own payment rates. Any reduction in payment that results from the MMA may result in a similar reduction in payments from nongovernmental payers.
39
On February 17, 2009, President Obama signed into law The American Recovery and Reinvestment Act of 2009. This law provides funding for the federal government to compare the effectiveness of different treatments for the same illness. This research is overseen by the Department of Health and Human Services, the Agency for Healthcare Research and Quality and the National Institutes for Health, and periodic reports on the status of the research and related expenditures must be made to Congress. Although the results of the comparative effectiveness studies are not intended to mandate coverage policies for public or private payers, it is not clear how such a result could be avoided and what if any effect the research will have on the sales of our product candidates, if any such product or the condition that it is intended to treat is the subject of a study. It is also possible that comparative effectiveness research demonstrating benefits in a competitor’s product could adversely affect the sales of our product candidates. Decreases in third-party reimbursement for our product candidates or a decision by a third-party payer to not cover our product candidates could reduce physician usage of the product candidate and have a material adverse effect on our sales, results of operations and financial condition.
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act of 2010 (collectively, the “ACA”) was enacted in March 2010. The ACA was enacted with the goal of expanding coverage for the uninsured while at the same time containing overall health care costs. With regard to pharmaceutical products, among other things, the ACA expanded and increased industry rebates for drugs covered under Medicaid programs and made changes to the coverage requirements under the Medicare D program. We still cannot fully predict the impact of the ACA on pharmaceutical companies as many of the ACA reforms require the promulgation of detailed regulations implementing the statutory provisions which has not yet been completed, and the Centers for Medicare & Medicaid Services has publicly announced that it is analyzing the ACA regulations and policies that have been issued to determine if changes should be made. In addition, although the U.S. Supreme Court has upheld the constitutionality of most of the ACA, some states have stated their intentions to not implement certain sections of the ACA and some members of Congress are still working to repeal the ACA. These challenges add to the uncertainty of the changes enacted as part of the ACA. In addition, the current legal challenges to the ACA, as well as Congressional efforts to repeal the ACA, add to the uncertainty of the legislative changes enacted as part of the ACA.
In addition, in some foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, some EU jurisdictions operate positive and negative list systems under which products may only be marketed once a reimbursement price has been agreed. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical trials that compare the cost-effectiveness of a particular product candidate to currently available therapies. Other member states allow companies to fix their own prices for medicines but monitor and control company profits. Such differences in national pricing regimes may create price differentials between EU member states. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our products. Historically, products launched in the EU do not follow price structures of the U.S. In the EU, the downward pressure on healthcare costs in general, particularly prescription medicines, has become intense. As a result, barriers to entry of new products are becoming increasingly high and patients are unlikely to use a drug product that is not reimbursed by their government or other public or private payers.
Other Health Care Laws and Compliance Requirements
In the U.S., our activities are potentially subject to regulation by various federal, state and local authorities in addition to the FDA, including the CMS, other divisions of the U.S. Department of Health and Human Services (e.g., the Office of Inspector General), the U.S. Department of Justice and individual U.S. Attorney offices within the Department of Justice, and state and local governments. For example, sales, marketing and scientific/educational grant programs must comply with the anti-fraud and abuse provisions of the Social Security Act, the False Claims Act, the privacy provisions of the Health Insurance Portability and Accountability Act, and similar state laws, each as amended. Pricing and rebate programs must comply with the Medicaid rebate requirements of the Omnibus Budget Reconciliation Act of 1990 and the Veterans Health Care Act of 1992 (“VHCA”), each as amended. If products are made available to authorized users of the federal supply schedule, additional laws and requirements apply. Under the VHCA, drug companies are required to offer certain drugs at a reduced price to a number of federal agencies including the U.S. Department of Veteran Affairs and U.S. Department of Defense, the Public Health Service and certain private Public Health Service-designated entities in order to participate in other federal funding programs including Medicare and Medicaid. In addition, discounted prices must be offered for certain U.S. Department of Defense purchases for its TRICARE program via a rebate system. Participation under the VHCA requires submission of pricing data and calculation of discounts and rebates pursuant to complex statutory formulas, as well as the entry into government procurement contracts governed by the federal acquisition regulations.
40
In order to distribute products commercially, we must comply with state laws that require the registration of manufacturers and wholesale distributors of pharmaceutical products in a state, including, in certain states, manufacturers and distributors who ship products into the state, even if such manufacturers or distributors have no place of business within the state. Several states have enacted legislation requiring pharmaceutical companies to establish marketing compliance programs, file periodic reports with the state, make periodic public disclosures on sales and marketing activities or register their sales representatives. Other legislation has been enacted in certain states prohibiting certain other sales and marketing practices. All of our activities are potentially subject to federal and state consumer protection and unfair competition laws. Likewise, these activities are subject to authorization or license requirements, or other legal requirements, under EU or EU member states’ law, or the law of other countries where we operate or have products manufactured or distributed.
Cost of Compliance with Environmental Laws
Our operations are subject to regulations under various federal, state, local and foreign laws concerning the environment, including laws addressing the discharge of pollutants into the air and water, the management and disposal of hazardous substances and wastes, and the cleanup of contaminated sites. We could incur substantial costs, including cleanup costs, fines and civil or criminal sanctions and third-party damage or personal injury claims, if in the future we were to violate or become liable under environmental laws. We are not aware of any costs or effects of our compliance with environmental laws.
Climate Change Related Regulation
Our operations are focused on research and development of pharmaceutical products, and a significant portion of such research and development is conducted outside of our facilities and by outsourced contract research organizations or universities. As a result, we do not anticipate any regulation surrounding climate change to impact our operations.
However, there is potential for more frequent and severe weather events and water availability challenges that may impact the facilities of our partners and our future suppliers. We cannot provide assurance that physical risks to the facilities of our partners and future suppliers and supply chain due to climate change will not occur in the future. We periodically review our vulnerability to potential weather-related risks and other natural disasters and update our assessments accordingly. Based on our reviews, we do not believe these potential risks are material to our operations at this time.
Employees and Human Capital Management
As of March 28, 2022, we and our subsidiaries had seven full-time employees. Two of these employees are located in the U.K., and five are located in the U.S.
In addition, we employ a limited number of part-time employees on a temporary basis, as well as scientific advisors, consultants and service providers, mainly through academic institutions and contract research organizations.
We have never had a work stoppage and none of our employees are covered by collective bargaining agreements or represented by a labor union. We believe that we have good relationships with our employees.
41
Our human capital resources objectives include, as applicable, identifying, recruiting, retaining, incentivizing, and integrating our existing and new employees, advisors, and consultants. The principal purposes of our equity and cash incentive plans are to attract, retain and reward personnel through the granting of stock-based and cash-based compensation awards, in order to increase stockholder value and the success of our company by motivating such individuals to perform to the best of their abilities and achieve our objectives.
In response to the COVID-19 pandemic, we focused our health and safety efforts on protecting our employees and their families. We swiftly implemented changes that we determined were in the best interest of our employees and the communities in which we operate, and which are aligned with guidance from the Centers for Disease Control and Prevention and in compliance with state and local regulations. This includes having all of our employees work from home.
COVID-19
In December 2019, a novel strain of coronavirus, which causes the infectious disease known as COVID-19, was reported in Wuhan, China. The World Health Organization declared COVID-19 a “Public Health Emergency of International Concern” on January 30, 2020 and a global pandemic on March 11, 2020. The effects of the COVID-19 pandemic and the responses to combat such pandemic have included government-mandated closures, stay-at-home orders and other related measures, the majority of which have since expired, but which, together with COVID-19 itself, have significantly impacted global economic activity and business investment in general, including causing supply chain issues and continuing disruptions in the labor markets. A continuation or worsening of the levels of market disruption and volatility seen in the recent past could have an adverse effect on our ability to access capital, and on our business, results of operations and financial condition. We have been closely monitoring the developments and have taken active measures to protect the health of our employees, their families, and our communities. The ultimate impact of the pandemic on us, our operations and prospects, will depend heavily on the continued duration of the COVID-19 pandemic and public health responses, including, vaccine availability and efficacy, the willingness of individuals to be vaccinated and to obtain booster shots, and virus mutations and seasonal outbreaks, as well as the substance and pace of macroeconomic recovery, all of which are uncertain and difficult to predict at this time. The follow up time for patient data and the statistical analysis for the Phase 2b Dupuytren’s Contracture clinical trial was delayed as a result of COVID-19, but such follow-up and statistical analysis are now completed and the Company announced the top-line data results from the Phase 2b trial on December 1, 2021. Additionally COVID-19 has delayed the initiation of certain clinical trials and may delay the initiation of other clinical trials in the future or otherwise have a material adverse effect on our future operations.
The pandemic is continuing and the full extent to which COVID-19 will ultimately impact us depends on future unknowable developments, including the duration and spread of the virus, the efficacy, availability and willingness of individuals to take vaccines, as well as potential new seasonal outbreaks.
We plan to continue to evaluate our business operations based on new information as it becomes available regarding the pandemic and will make changes that we consider necessary in light of any new developments.
Corporate History
Formation
We were formed as a blank check company organized under the laws of the State of Delaware on September 7, 2016. We were formed for the purpose of effecting a merger, capital stock exchange, stock purchase, asset acquisition or other similar business combination with one or more operating businesses. Since formation, we focused our efforts on acquiring an operating company in the healthcare and related wellness industry although our efforts in identifying a prospective target business were not limited to a particular industry.
42
Initial Public Offering
On June 7, 2017, pursuant to the Company’s Initial Public Offering (the “IPO”), the Company sold 11,500,000 Units at a purchase price of $10.00 per Unit, inclusive of 1,500,000 Units sold to the underwriters on June 23, 2017 upon the underwriters’ election to fully exercise their over-allotment option, generating gross proceeds of $115,000,000. Each “Unit” consisted of one share of the Company’s common stock, one right to receive one-tenth of one share of the Company’s common stock upon the consummation of a business combination (“Right”), and one redeemable warrant to purchase one-half of one share of the Company’s common stock (the “Public Warrants”). Each Public Warrant entitles the holder to purchase one-half of one share of common stock at an exercise price of $5.75 per half share ($11.50 per whole share), subject to adjustment. No fractional shares will be issued upon exercise of the Public Warrants. The Public Warrants became exercisable 12 months from the closing of the IPO, and expire five years after the completion of the Business Combination.
The Company may redeem the Public Warrants, in whole and not in part, at a price of $0.01 per Public Warrant upon 30 days’ notice (“30-day redemption period”), only in the event that the last sale price of the common stock equals or exceeds $18.00 per share for any 20 trading days within a 30-trading day period ending on the third trading day prior to the date on which notice of redemption is given, provided there is an effective registration statement with respect to the shares of common stock underlying such Public Warrants and a current prospectus relating to those shares of common stock is available throughout the 30-day redemption period. If the Company calls the Public Warrants for redemption as described above, the Company’s management will have the option to require all holders that wish to exercise Public Warrants to do so on a “cashless basis.” In determining whether to require all holders to exercise their Public Warrants on a “cashless basis,” management will consider, among other factors, the Company’s cash position, the number of Public Warrants that are outstanding and the dilutive effect on the Company’s stockholders of issuing the maximum number of shares of common stock issuable upon the exercise of the Public Warrants. Each holder of a Right received one-tenth (1/10) of one share of common stock upon consummation of the Business Combination. No fractional shares were issued upon exchange of the Rights.
Private Placement
Concurrent with the closing of the IPO, KBL IV Sponsor LLC (the “Sponsor”) and the underwriters purchased an aggregate of 450,000 unregistered Units (“Private Units”) at $10.00 per Unit, generating gross proceeds of $4,500,000 in a private placement. In addition, on June 23, 2017, the Company consummated the sale of an additional 52,500 Private Units at a price of $10.00 per Unit, which were purchased by the Sponsor and underwriters, generating gross proceeds of $525,000. Of these, 377,500 Private Units were purchased by the Sponsor and 125,000 Private Units were purchased by the underwriters. The proceeds from the Private Units were added to the net proceeds from the IPO held in a Trust Account (the “Trust Account”). The Private Units (including their component securities) were not transferable, assignable or salable until 30 days after the completion of the Business Combination and the warrants included in the Private Units (the “Private Placement Warrants”) will be non-redeemable so long as they are held by the Sponsor, the underwriters or their permitted transferees. If the Private Placement Warrants are held by someone other than the Sponsor, the underwriters or their permitted transferees, the Private Placement Warrants will be redeemable by the Company and exercisable by such holders on the same basis as the warrants included in the Units sold in the IPO. In addition, for as long as the Private Placement Warrants are held by the underwriters or its designees or affiliates, they may not be exercised after five years from the effective date of the registration statement related to the IPO. Otherwise, the Private Placement Warrants have terms and provisions that are identical to those of the warrants sold as part of the Units in the IPO and have no net cash settlement provisions.
43
Business Combination
On July 25, 2019, we entered into a Business Combination Agreement (as amended from time to time, the “Business Combination Agreement”), with KBL Merger Sub, Inc. (“Merger Sub”), 180 Life Corp. (f/k/a 180 Life Sciences Corp.)(“180”), Katexco Pharmaceuticals Corp. (“Katexco”), CannBioRex Pharmaceuticals Corp. (“CBR Pharma”), 180 Therapeutics L.P. (“180 LP” and together with Katexco and CBR Pharma, the “180 Subsidiaries” and, together with 180 Life Sciences Corp., the “180 Parties”), and Lawrence Pemble, in his capacity as representative of the stockholders of the 180 Parties (the “Stockholder Representative”). The business combination described in the Business Combination Agreement (the “Business Combination”), closed and became effective on November 6, 2020 (the “Closing”). Pursuant to the Business Combination Agreement, among other things, Merger Sub merged with and into 180, with 180 continuing as the surviving entity and a wholly-owned subsidiary of the Company (the “Merger”). In connection with, and prior to, the Closing, 180 Life Sciences Corp. filed a Certificate of Amendment of its Certificate of Incorporation in Delaware to change its name to 180 Life Corp., and our company (which was known as of our entry into the Business Combination as KBL Merger Corp. IV, changed our name to 180 Life Sciences Corp.).
180 was incorporated in Delaware on January 28, 2019. Prior to the Closing of the Business Combination, 180 operated through three subsidiaries: 180 LP, a Delaware limited partnership formed on September 6, 2013; Katexco, a company incorporated in British Columbia, Canada on March 7, 2018; and CBR Pharma, a company incorporated in British Columbia, Canada on March 8, 2018.
In July 2019, 180 and each of 180 LP, Katexco and CBR Pharma completed a corporate restructuring, pursuant to which 180 LP, Katexco and CBR Pharma became wholly-owned subsidiaries of 180LS (the “Reorganization”). The corporate restructuring arrangements with respect to Katexco and CBR Pharma were completed under the Business Corporations Act (British Columbia).
On November 6, 2020 (the “Closing Date”), the Company consummated the Business Combination following a special meeting of stockholders held on November 5, 2020, where the stockholders of the Company considered and approved, among other matters, a proposal to adopt the Business Combination. Pursuant to the Business Combination Agreement, among other things, Merger Sub merged with and into 180, with 180 continuing as the surviving entity and a wholly-owned subsidiary of the Company. The Merger became effective on November 6, 2020 (such time, the “Effective Time”, and the closing of the Merger being referred to herein as the “Closing”). In connection with, and prior to, the Closing, 180 filed a Certificate of Amendment of its Certificate of Incorporation in Delaware to change its name to 180 Life Corp. and KBL Merger Corp. IV changed its name to 180 Life Sciences Corp.
At the Effective Time, each share of 180 common stock issued and outstanding prior to the Effective Time was automatically converted into the right to receive approximately 168.3784 shares of the common stock, par value $0.0001 per share, of the Company (such shares of Common Stock issuable to the common stockholders of 180 pursuant to the Business Combination Agreement, the “Merger Consideration Shares”). An aggregate of 17,494,725 shares of common stock have been issued to date to the common stockholders of 180 as Merger Consideration Shares, including the Escrow Shares (as defined below). Also at the Effective Time, each share underlying the 180 preferred stock issued and outstanding prior to the Effective Time was converted into the right to receive one Class C Special Voting Share of the Company, or one Class K Special Voting Share of the Company, as applicable (such shares, the “Special Voting Shares”). The Special Voting Shares entitle the holder thereof to an aggregate number of votes, on any particular matter, proposition or question, equal to the number of Exchangeable Shares (as defined below) of each of CannBioRex Purchaseco ULC and Katexco Purchaseco ULC, Canadian subsidiaries of 180, respectively, that are outstanding from time to time.
As a result of the Merger, the existing exchangeable shares (collectively, the “Exchangeable Shares”) of CannBioRex Purchaseco ULC and/or Katexco Purchaseco ULC were adjusted in accordance with the share provisions in the articles of CannBioRex Purchaseco ULC or Katexco Purchaseco ULC, as applicable, governing the Exchangeable Shares such that they were multiplied by the exchange ratio for the Merger and became exchangeable into shares of Common Stock. The Exchangeable Shares entitle the holders to dividends and other rights that are substantially economically equivalent to those of holders of Common Stock, and holders of Exchangeable Shares have the right to vote at meetings of the stockholders of the Company. An aggregate of 5,275 shares of Common Stock are reserved for issuance to the holders of the Exchangeable Shares upon the exchange thereof.
44
Pursuant to the Business Combination Agreement, 1,049,999 of the Merger Consideration Shares (such shares, the “Escrow Shares”) were deposited into an escrow account (the “Escrow Account”) to serve as security for, and the exclusive source of payment of, the Company’s indemnity rights under the Business Combination Agreement, all of which will be released to the same stockholders 12 months following the Closing of the Business Combination.
As a result of the Business Combination, the former stockholders of 180 became the controlling stockholders of the Company and 180 became a wholly-owned subsidiary of the Company. The Business Combination was accounted for as a reverse merger, whereby 180 is considered the acquirer for accounting and financial reporting purposes.
In connection with the Closing, the Company withdrew $9,006,493 of funds from the Trust Account (as defined below) to fund the redemptions of 816,461 shares.
The chart below shows our current organizational structure:
45
About Us
Our principal executive offices are located at 3000 El Camino Real, Bldg. 4, Suite 200, Palo Alto, CA 94306, and our telephone number is (650) 507-0669. We maintain a website at www.180lifesciences.com. We have not incorporated by reference into this Report the information in, or that can be accessed through, our website, and you should not consider it to be a part of this Report.
Jumpstart Our Business Startups Act
In April 2012, the Jumpstart Our Business Startups Act (“JOBS Act”) was enacted into law. The JOBS Act provides, among other things:
| ● | Exemptions for “emerging growth companies” from certain financial disclosure and governance requirements for up to five years and provides a new form of financing to small companies; | |
| ● | Amendments to certain provisions of the federal securities laws to simplify the sale of securities and increase the threshold number of record holders required to trigger the reporting requirements of the Exchange Act; | |
| ● | Relaxation of the general solicitation and general advertising prohibition for Rule 506 offerings; | |
| ● | Adoption of a new exemption for public offerings of securities in amounts not exceeding $50 million; and | |
| ● | Exemption from registration by a non-reporting company of offers and sales of securities of up to $1,000,000 that comply with rules to be adopted by the SEC pursuant to Section 4(6) of the Securities Act and exemption of such sales from state law registration, documentation or offering requirements. | |
In general, under the JOBS Act a company is an “emerging growth company” if its initial public offering (“IPO”) of common equity securities was affected after December 8, 2011 and the company had less than $1.07 billion of total annual gross revenues during its last completed fiscal year. A company will no longer qualify as an “emerging growth company” after the earliest of
| (i) | the completion of the fiscal year in which the company has total annual gross revenues of $1.07 billion or more, |
| (ii) | the completion of the fiscal year of the fifth anniversary of the company’s IPO (December 31, 2022); |
| (iii) | the company’s issuance of more than $1 billion in nonconvertible debt in the prior three-year period, or |
| (iv) | the company becoming a “large accelerated filer” as defined under the Exchange Act. |
The JOBS Act provides additional new guidelines and exemptions for non-reporting companies and for non-public offerings. Those exemptions that impact the Company are discussed below.
Financial Disclosure. The financial disclosure in a registration statement filed by an “emerging growth company” pursuant to the Securities Act, will differ from registration statements filed by other companies as follows:
| (i) | audited financial statements required for only two fiscal years (provided that “smaller reporting companies” such as the Company are only required to provide two years of financial statements); |
| (ii) | selected financial data required for only the fiscal years that were audited (provided that “smaller reporting companies” such as the Company are not required to provide selected financial data as required by Item 301 of Regulation S-K); and |
46
| (iii) | executive compensation only needs to be presented in the limited format now required for “smaller reporting companies”. |
However, the requirements for financial disclosure provided by Regulation S-K promulgated by the Rules and Regulations of the SEC already provide certain of these exemptions for smaller reporting companies. The Company is a smaller reporting company. Currently a smaller reporting company is not required to file as part of its registration statement selected financial data and only needs to include audited financial statements for its two most current fiscal years with no required tabular disclosure of contractual obligations.
The JOBS Act also exempts the Company’s independent registered public accounting firm from having to comply with any rules adopted by the Public Company Accounting Oversight Board (“PCAOB”) after the date of the JOBS Act’s enactment, except as otherwise required by SEC rule.
The JOBS Act further exempts an “emerging growth company” from any requirement adopted by the PCAOB for mandatory rotation of the Company’s accounting firm or for a supplemental auditor report about the audit.
Internal Control Attestation. The JOBS Act also provides an exemption from the requirement of the Company’s independent registered public accounting firm to file a report on the Company’s internal control over financial reporting, although management of the Company is still required to file its report on the adequacy of the Company’s internal control over financial reporting.
Section 102(a) of the JOBS Act exempts “emerging growth companies” from the requirements in §14A(e) of the Exchange Act for companies with a class of securities registered under the Exchange Act to hold stockholder votes for executive compensation and golden parachutes.
Other Items of the JOBS Act. The JOBS Act also provides that an “emerging growth company” can communicate with potential investors that are qualified institutional buyers or institutions that are accredited to determine interest in a contemplated offering either prior to or after the date of filing the respective registration statement. The JOBS Act also permits research reports by a broker or dealer about an “emerging growth company” regardless of whether such report provides sufficient information for an investment decision. In addition, the JOBS Act precludes the SEC and FINRA from adopting certain restrictive rules or regulations regarding brokers, dealers and potential investors, communications with management and distribution of research reports on the “emerging growth company’s” initial public offerings (IPOs).
Section 106 of the JOBS Act permits “emerging growth companies” to submit registration statements under the Securities Act on a confidential basis provided that the registration statement and all amendments thereto are publicly filed at least 15 days before the issuer conducts any road show. This is intended to allow “emerging growth companies” to explore the IPO option without disclosing to the market the fact that it is seeking to go public or disclosing the information contained in its registration statement until the company is ready to conduct a roadshow.
Election to Opt Out of Transition Period. Section 102(b)(1) of the JOBS Act exempts “emerging growth companies” from being required to comply with new or revised financial accounting standards until private companies (that is, those that have not had a Securities Act registration statement declared effective or do not have a class of securities registered under the Exchange Act) are required to comply with the new or revised financial accounting standard.
The JOBS Act provides that a company can elect to opt out of the extended transition period and comply with the requirements that apply to non-emerging growth companies but any such election to opt out is irrevocable. The Company has elected not to opt out of the transition period.
47
Summary Risk Factors
We face risks and uncertainties related to our business, many of which are beyond our control. In particular, risks associated with our business include:
| ● | we are a clinical stage biotechnology company that had no revenue for the years ended December 31, 2021 and 2020, and do not anticipate generating revenue for the near future; |
| ● | our need for additional financing, both near term and long term, to support our operations, our ability to raise such financing as needed, the terms of such financing, if available, potential significant dilution associated therewith, and covenants and restrictions we may need to comply with in connection with such funding; |
| ● | our dependence on the success of our future product candidates, some of which may not receive regulatory approval or be successfully commercialized; problems in our manufacturing process for our new products and/or our failure to comply with manufacturing regulations, or unexpected increases in our manufacturing costs; problems with distribution of our products; and failure to adequately market our products; |
| ● | risks associated with the growth of our business, our ability to maintain such growth, difficulties in managing our growth, and executing our growth strategy; |
| ● | liability for previously restated financial statements and associated with ineffective controls and procedures; |
| ● | our dependence on our key personnel and our ability to attract and retain employees; |
| ● | risks from intense competition from companies with greater resources and experience than we have; |
| ● | risks that our future product candidates, if approved, may be unable to achieve the expected market acceptance and, consequently, limit our ability to generate revenue from new products; |
| ● | the outcome of currently pending and future claims and litigation, future government investigations, and other proceedings may adversely affect our business and results of operations; |
| ● | the fact that the majority of our license agreements provide the licensors and/or counter-parties the right to use and/or exploit such licensed intellectual property; |
| ● | preclinical studies and earlier clinical trials may not necessarily be predictive of future results and may not have favorable results; we have limited marketing experience, and our future ability to successfully commercialize any of our product candidates, even if they are approved in the future is unknown; and business interruptions could delay us in the process of developing our future product candidates and could disrupt our product sales; |
| ● | third-party payors may not provide coverage and adequate reimbursement levels for any future products; |
| ● | liability from lawsuits (including product liability lawsuits, stockholder lawsuits and regulatory matters), including judgments, damages, fines and penalties and including the outcome of currently pending litigation, potential future government investigations, and other proceedings that may adversely affect our business and results of operations; |
| ● | security breaches, loss of data and other disruptions which could prevent us from accessing critical information or expose us to liabilities or damages; |
48
| ● | risks associated with clinical trials that are expensive, time-consuming, uncertain and susceptible to change, delay or termination and which are open to differing interpretations; |
| ● | our ability to comply with existing and future rules and regulations, including federal, state and foreign healthcare laws and regulations and implementation of, or changes to, such healthcare laws and regulations; |
| ● | delays in the trials, testing, application, or approval process for drug candidates and/or our ability to obtain approval for promising drug candidates, and the costs associated therewith; |
| ● | our ability to adequately protect our future product candidates or our proprietary technology in the marketplace, claims and liability from third parties regarding our alleged infringement of their intellectual property; |
| ● | differences in laws and regulations between countries and other jurisdictions; |
| ● | changes in laws or regulations, including, but not limited to tax laws and controlled substance laws, or a failure to comply with any laws and regulations; |
| ● | conflicts of interest between our officers, directors, consultants and scientists; |
| ● | penalties associated with our failure to comply with certain pre-agreed contractual obligations and restrictions; |
| ● | dilution caused by future fund raising, the conversion/exercise of outstanding convertible securities, and downward pressure on the value of our securities caused by such future issuances/sales; |
| ● | negative effects on our business from the COVID-19 pandemic and other potential future pandemics; |
| ● | the extremely volatile nature of our securities and potential lack of liquidity therefore; |
| ● | the fact that our Certificate of Incorporation provides for indemnification of officers and directors, limits the liability of officers and directors, allows for the authorization of preferred stock without stockholder approval, and includes certain anti-takeover provisions; |
| ● | our ability to maintain the listing of our common stock and warrants on NASDAQ and the costs of compliance with SEC and NASDAQ rules and requirements; |
| ● | risks associated with our status as an emerging growth company and the provisions of the JOBS Act, which we are able to take advantage of, due to such status; |
| ● | failure of our information technology systems, including cybersecurity attacks or other data security incidents, that could significantly disrupt the operation of our business; |
| ● | the fact that we may acquire other companies which could divert our management’s attention, result in additional dilution to our stockholders and otherwise disrupt our operations and harm our operating results and if we make any acquisitions, they may disrupt or have a negative impact on our business; |
| ● | the fact that we may apply working capital and future funding to uses that ultimately do not improve our operating results or increase the value of our securities; and |
| ● | our growth depends in part on the success of our strategic relationships with third parties. |
49
You should be aware that there are substantial risks for an investment in our common stock. You should carefully consider these risk factors before you decide to invest in our common stock.
If any of the following risks were to occur, our business, financial condition, results of operations or other prospects, could be materially adversely effected, and the occurrence of any of these risks could materially affect our likelihood of success. If that happens, the market price of our common stock, if any, could decline, and prospective investors would lose all or part of their investment in our common stock.
Risks Related to Our Business Operations
Our business, financial condition and results of operations are subject to various risks and uncertainties, including those described below. This section discusses factors that, individually or in the aggregate, could cause our actual results to differ materially from expected and historical results. Our business, financial condition or results of operations could be materially adversely affected by any of these risks. It is not possible to predict or identify all such factors. Consequently, the following description of Risk Factors is not a complete discussion of all potential risks or uncertainties applicable to our business.
Our current cash balance is only sufficient to fund our planned business operations through the third quarter of 2022. If additional capital is not available, we may not be able to pursue our planned business operations, may be forced to change our planned business operations, or may take other actions that could adversely impact our stockholders.
We are a clinical stage biotechnology company that currently has no revenue. Thus, our business does not generate the cash necessary to finance our planned business operations. We will require significant additional capital to: (i) develop FDA-approved products and commercialize such products; (ii) fund research and development activities relating to, and obtain regulatory approval for, our product candidates; (iii) protect our intellectual property; (iv) attract and retain highly-qualified personnel; (v) respond effectively to competitive pressures; and (vi) acquire complementary businesses or technologies.
Our future capital needs depend on many factors, including: (i) the scope, duration and expenditures associated with our research, development and commercialization efforts; (ii) continued scientific progress in our programs; (iii) the outcome of potential partnering or licensing transactions, if any; (iv) competing technological developments; (v) our proprietary patent position; and (vi) the regulatory approval process for our products.
We will need to raise substantial additional funds through public or private equity offerings, debt financings or strategic alliances and licensing arrangements to finance our planned business operations. We may not be able to obtain additional financing on terms favorable to us, if at all. General market conditions, as well as the ongoing COVID-19 pandemic, may make it difficult for us to seek financing from the capital markets, and the terms of any financing may adversely affect the holdings or the rights of our stockholders. For example, if we raise additional funds by issuing equity securities, further dilution to our stockholders will result, which may substantially dilute the value of their investment. Any equity financing may also have the effect of reducing the conversion or exercise price of our outstanding convertible or exercisable securities, which could result in the issuance (or potential issuance) of a significant number of additional shares of our common stock. In addition, as a condition to providing additional funds to us, future investors may demand, and may be granted, rights superior to those of existing stockholders. Debt financing, if available, may involve restrictive covenants that could limit our flexibility to conduct future business activities and, in the event of insolvency, could be paid before holders of equity securities received any distribution of our assets. We may be required to relinquish rights to our technologies or product candidates, or grant licenses through alliance, joint venture or agreements on terms that are not favorable to us, in order to raise additional funds. If adequate funds are not available, we may have to delay, reduce or eliminate one or more of our planned activities with respect to our business, or terminate our operations. These actions would likely reduce the market price of our common stock.
50
We will need additional capital which may not be available on commercially acceptable terms, if at all, which raises questions about our ability to continue as a going concern.
As of December 31, 2021, we had an accumulated deficit of $68,682,286, net loss amounted to $20,324,648 and we had a working capital deficit of $8,498,193. As of March 28, 2022, we had cash on hand of $6,045,848. The accompanying consolidated financial statements have been prepared assuming the Company will continue as a going concern. As we are not generating revenues, we need to raise a significant amount of capital in order to pay our debts and cover our operating costs. While the Company raised money in August 2021, there is no assurance that we will be able to raise additional needed capital or that such capital will be available under favorable terms.
We are subject to all the substantial risks inherent in the development of a new business enterprise within an extremely competitive industry. Due to the absence of a long-standing operating history and the emerging nature of the markets in which we compete, we anticipate operating losses until we can successfully implement our business strategy, which includes all associated revenue streams. We may never ever achieve profitable operations or generate significant revenues.
We believe that in the aggregate, we will require significant additional capital funding to support and expand the research and development and marketing of our products, fund future clinical trials, repay debt obligations, provide capital expenditures for additional equipment and development costs, payment obligations, office space and systems for managing the business, and cover other operating costs until our planned revenue streams from products are fully-implemented and begin to offset our operating costs, if ever.
Since our inception, we have funded our operations with the proceeds from equity and debt financings. We have experienced liquidity issues due to, among other reasons, our limited ability to raise adequate capital on acceptable terms. We have historically relied upon the issuance of equity and promissory notes that are convertible into shares of our common stock to fund our operations and have devoted significant efforts to reduce that exposure. We anticipate that we will need to issue equity to fund our operations and fund our operating expenses for the foreseeable future. If we are unable to achieve operational profitability or we are not successful in securing other forms of financing, we will have to evaluate alternative actions to reduce our operating expenses and conserve cash.
These conditions raise substantial doubt about our ability to continue as a going concern for the next twelve months. The accompanying consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. Accordingly, the consolidated financial statements do not include any adjustments relating to the recoverability of assets and classification of liabilities that might be necessary should the Company be unable to continue as a going concern. The consolidated financial statements included herein also include a going concern footnote.
Additionally, wherever possible, our Board of Directors will attempt to use non-cash consideration to satisfy obligations. In many instances, we believe that the non-cash consideration will consist of restricted shares of our common stock, preferred stock or warrants to purchase shares of our common stock. Our Board of Directors has authority, without action or vote of the shareholders, but subject to NASDAQ rules and regulations (which generally require shareholder approval for any transactions which would result in the issuance of more than 20% of our then outstanding shares of common stock or voting rights representing over 20% of our then outstanding shares of stock), to issue all or part of the authorized but unissued shares of common stock, preferred stock or warrants to purchase such shares of common stock. In addition, we may attempt to raise capital by selling shares of our common stock, possibly at a discount to market in the future. These actions will result in dilution of the ownership interests of existing shareholders, may further dilute common stock book value, and that dilution may be material. Such issuances may also serve to enhance existing management’s ability to maintain control of us, because the shares may be issued to parties or entities committed to supporting existing management.
51
We have significant and increasing liquidity needs and may require additional funding.
Research and development, management and administrative expenses, including legal expenses, and cash used for operations will continue to be significant and may increase substantially in the future in connection with new research and development initiatives, clinical trials, continued product commercialization efforts and the launch of our future product candidates. We will need to raise additional capital to fund our operations, continue to conduct clinical trials to support potential regulatory approval of marketing applications, and to fund commercialization of our future product candidates.
The amount and timing of our future funding requirements will depend on many factors, including, but not limited to:
| ● | the timing of FDA approval, if any, and approvals in international markets of our future product candidates, if at all; |
| ● | the timing and amount of revenue from sales of our products, or revenue from grants or other sources; |
| ● | the rate of progress and cost of our clinical trials and other product development programs; |
| ● | costs of establishing or outsourcing sales, marketing and distribution capabilities; |
| ● | costs and timing of any outsourced growing and commercial manufacturing supply arrangements for our future product candidates; |
| ● | costs of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights associated with our future product candidates; |
| ● | the effect of competing technological and market developments; |
| ● | personnel, facilities and equipment requirements; and |
| ● | the terms and timing of any additional collaborative, licensing, co-promotion or other arrangements that we may establish. |
While we expect to fund our future capital requirements from a number of sources, such as cash flow from operations and the proceeds from further public and/or private offerings, we cannot assure you that any of these funding sources will be available to us on favorable terms, or at all. Further, even if we can raise funds from all of the above sources, the amounts raised may not be sufficient to meet our future capital requirements.
We are dependent on the success of our future product candidates, some of which may not receive regulatory approval or be successfully commercialized.
Our success will depend on our ability to successfully develop and commercialize our future product candidates through our development programs, including our product candidate for the treatment of Dupuytren’s Contracture and any other product candidates developed through our fibrosis & anti-TNF, CBD derivatives, and α7nAChR development platforms. We may never be able to develop products which receive regulatory approval in the U.S. or elsewhere. There can be no assurance that the FDA, EMA or any other regulatory authority will approve these product candidates.
Our ability to successfully commercialize our future product candidates will depend on, among other things, our ability to successfully complete pre-clinical and other non-clinical studies and clinical trials and to receive regulatory approvals from the FDA and similar foreign regulatory authorities. Delays in the regulatory process could have a material adverse effect on our business, results of operations and financial condition.
52
We have recently grown our business and will need to increase the size and complexity of our organization in the future, and we may experience difficulties in managing our growth and executing our growth strategy.
Our management, personnel and systems currently in place may not be adequate to support our business plan and future growth. We will need to increase our number of full-time equivalent employees in order to conduct Phase 1, 2 and 3 clinical trials of our future products and to establish a commercial organization and commercial infrastructure. As a result of these future activities, the complexity of our business operations is expected to substantially increase. We will need to develop and expand our scientific, manufacturing, sales and marketing, managerial, compliance, operational, financial and other resources to support our planned research, development, manufacturing and commercialization activities.
Our need to effectively manage our operations, growth and various projects requires that we:
| ● | continue to improve our operational, financial, management and regulatory compliance controls and reporting systems and procedures; |
| ● | attract and retain sufficient numbers of talented employees; |
| ● | manage our commercialization activities effectively and in a cost-effective manner (currently trial and development for our clinical trials is very cost effective); and |
| ● | manage our development efforts effectively while carrying out our contractual obligations to contractors and other third parties. |
We have utilized and continue to utilize the services of part-time outside consultants and contractors to perform a number of tasks for our company, including tasks related to compliance programs, clinical trial management, regulatory affairs, formulation development and other drug development functions. Our growth strategy may entail expanding our use of consultants and contractors to implement these and other tasks going forward. If we are not able to effectively expand our organization by hiring new employees and expanding our use of consultants and contractors, we may be unable to successfully implement the tasks necessary to effectively execute on our planned research, development, manufacturing and commercialization activities and, accordingly, may not achieve our research, development and commercialization goals.
We face liability for previously restated financial statements.
We filed a Current Report on Form 8-K on December 31, 2020 and another Current Report on Form 8-K on February 3, 2021, where we announced that due to matters we discovered which related to KBL, prior to the Business Combination, certain historical financial statements were unreliable. As a result, we restated our financial statements for the three and six months ended June 30, 2020 and for the three and nine months ended September 30, 2020, because of errors in such financial statements which were identified after such financial statements were filed with the SEC in our original quarterly reports for the quarters ended June 30, 2020 and September 30, 2020. While we believe these restatements are the result of the actions of, and are the responsibility of, the management of KBL (none of whom remain employed by the Company), we may be subject to stockholder litigation, rating downgrades, negative publicity and difficulties in attracting and retaining key clients, employees and management personnel as a result of such restatements. Additionally, our securities may trade at prices lower than similarly situated companies which have not had to restate their financial statements.
Our failure to appropriately account for complex financial instruments may result in the requirement that we restate our financial statements.
Certain of our current securities, and future securities we issue may, require complex accounting treatment and analysis. The SEC recently issued a Statement on Accounting and Reporting Considerations for Warrants Issued by Special Purpose Acquisition Companies (“SPACs”)(the “Statement”) on April 12, 2021 and may in the future issue further statements on SPAC accounting. While we believe that our financial statements for the years ended December 31, 2021 and 2020, and for the quarters ended March 31, 2021, June 30, 2021 and September 30, 2021, comply with the guidance issued on April 12, 2021, it is possible that as a result of the information and guidance set forth in the Statement, or in future statements or advisories released by the SEC or an accounting standards board, that we will need to restate our financial statements, and such guidance (including as set forth in the Statement), could have a material adverse effect on our financial condition and results of operations for prior periods which are required to be restated, if any, and/or on future periods moving forward, even if a restatement is not required. We do not believe that the SEC’s prior guidance regarding the accounting treatment of redeemable shares issued by SPACs will require a restatement of our historical financial statements or will have any impact on the financial statements that we plan to prepare and file in the future.
53
Operating results may vary significantly in future periods.
Our financial results are unpredictable and may fluctuate, for among other reasons, due to commercial sales of our future product candidates; our achievement of product development objectives and milestones; clinical trial enrollment and expenses; research and development expenses; and the timing and nature of contract manufacturing and contract research payments. A high portion of our costs are predetermined on an annual basis, due in part to our significant research and development costs. Thus, small declines in future revenue could disproportionately affect financial results in a quarter.
We depend on our key personnel and our ability to attract and retain employees.
Our future growth and success depend on our ability to recruit, retain, manage and motivate our employees. We are highly dependent on our current management and scientific personnel, including our Chief Executive Officer, Dr. James N. Woody, our Co-Chairmen, Sir Marc Feldmann, Ph.D., and Lawrence Steinman, M.D., our Chief Scientific Officer, Jonathan Rothbard, Ph.D., our Chief Operating Officer / Chief Business Officer, Quan Anh Vu, and our scientists, Raphael Mechoulam and Jagdeep Nanchahal. The inability to hire or retain experienced management personnel could adversely affect our ability to execute our business plan and harm our operating results. Due to the specialized scientific and managerial nature of our business, we rely heavily on our ability to attract and retain qualified scientific, technical and managerial personnel. The competition for qualified personnel in the biotechnological field is intense and we may be unable to continue to attract and retain qualified personnel necessary for the development of our business or to recruit suitable replacement personnel.
Problems in our manufacturing process for our new chemical entities, failure to comply with manufacturing regulations or unexpected increases in our manufacturing costs could harm our business, results of operations and financial condition.
We are responsible for the manufacture and supply of our future product candidates in the CBD derivatives and α7nAChR programs for commercial use and for use in clinical trials. The manufacturing of our future product candidates necessitates compliance with GMPs and other regulatory requirements in international jurisdictions. Our ability to successfully manufacture our future product candidates will involve manufacture of finished products and labeling and packaging, which includes product information, tamper proof evidence and anti-counterfeit features, under tightly controlled processes and procedures. In addition, we will have to ensure chemical consistency among our batches, including clinical trial batches and, if approved, marketing batches. Demonstrating such consistency may require typical manufacturing controls as well as clinical data. We will also have to ensure that our batches conform to complex release specifications. If we are unable to manufacture our future product candidates in accordance with regulatory specifications, or if there are disruptions in our manufacturing process due to damage, loss or otherwise, or failure to pass regulatory inspections of our manufacturing facilities, we may not be able to meet demand or supply sufficient product for use in clinical trials, and this may also harm our ability to commercialize our future product candidates on a timely or cost-competitive basis, if at all.
We may not develop and expand our manufacturing capability in time to meet demand for our product candidates, and the FDA or foreign regulatory authorities may not accept our facilities or those of our contract manufacturers as being suitable for the production of our products and product candidates. Any problems in our manufacturing process could materially adversely affect our business, results of operations and financial condition.
54
Our memorandum of understanding with Celltrion Healthcare may not result in the parties entering into a definitive agreement.
In September 2021, we entered into a non-binding memorandum of understanding with Celltrion Healthcare, a biopharmaceutical company, for the supply of an anti-TNF biosimilar drug used in our ongoing development of anti-TNF products. The parties have not entered into a definitive agreement regarding such relationship to date, and such definitive agreement may not ultimately be entered into on terms contemplated, if at all. In the event that we are unable to come to mutually agreeable definitive terms with Celltrion Healthcare, we will need to locate an alternative supplier of the anti-TNF biosimilar drug, and we may be unable to find an alternative supplier or such alternative supplier may require less favorable terms than are currently contemplated. Any of the above may materially adversely affect our business, results of operations and financial condition.
We expect to face intense competition from companies with greater resources and experience than we have; and may face competition from competitors seeking to market our products under a Section 505(b)(2) application.
The pharmaceutical industry is highly competitive and subject to rapid change. The industry continues to expand and evolve as an increasing number of competitors and potential competitors enter the market. Many of these competitors and potential competitors have substantially greater financial, technological, managerial and research and development resources and experience than our company. Some of these competitors and potential competitors have more experience than our company in the development of pharmaceutical products, including validation procedures and regulatory matters. In addition, our future product candidates, if successfully developed, will compete with product offerings from large and well-established companies that have greater marketing and sales experience and capabilities than our company or our collaboration partners have. In particular, Insys Therapeutics, Inc. is developing CBD in Infantile Spasms (“IS”), and potentially other indications. Zogenix, Inc. has reported positive data in two Phase 3 trials of low dose fenfluramine in Dravet syndrome and has commenced a Phase 3 trial with this product in Lennox Gastaut Syndrome. Biocodex recently received regulatory approval from the FDA for the drug Stiripentol (Diacomit) for the treatment of Dravet syndrome. Other companies with greater resources than our company may announce similar plans in the future. In addition, there are non-FDA approved CBD preparations being made available from companies in the medical marijuana industry, which might attempt to compete with our future product candidates.
Additionally, competitors may also seek to market versions of our drug products via a section 505(b)(2) application, which is a type of somewhat abbreviated NDA. NDA Section 505(b)(2) applications may be submitted for drug products that represent a modification, such as a new indication or new dosage form, of a previously approved drug. Section 505(b)(2) applications may rely on the FDA’s previous findings for the safety and effectiveness of the previously approved drug in addition to information obtained by the 505(b)(2) applicant to support the modification of the previously approved drug. Preparing Section 505(b)(2) applications may be less costly and time-consuming than preparing an NDA based entirely on new data and information. Section 505(b)(2) applications are subject to the same patent certification procedures as an ANDA.
If we are unable to compete successfully, our commercial opportunities will be reduced and our business, results of operations and financial conditions may be materially harmed.
55
Our future product candidates, if approved, may be unable to achieve the expected market acceptance and, consequently, limit our ability to generate revenue from new products.
Even when product development is successful and regulatory approval has been obtained, our ability to generate sufficient revenue depends on the acceptance of our products by physicians and patients. We cannot assure you that any of our future product candidates will achieve the expected level of market acceptance and revenue if and when they obtain the requisite regulatory approvals. The market acceptance of any product depends on a number of factors, including the indication statement, warnings required by regulatory authorities in the product label and new competing products. Market acceptance can also be influenced by continued demonstration of efficacy and safety in commercial use, physicians’ willingness to prescribe the product, reimbursement from third-party payors such as government health care programs and private third-party payors, the price of the product, the nature of any post-approval risk management activities mandated by regulatory authorities, competition, and marketing and distribution support. Further, our U.S. distribution depends on the adequate performance of a reimbursement support hub and contracted specialty pharmacies in a closed-distribution network. An ineffective or inefficient U.S. distribution model at launch may lead to inability to fulfill demand, and consequently a loss of revenue. The success and acceptance of a product in one country may be negatively affected by its activities in another. If we fail to adapt our approach to clinical trials in the U.S. market to meet the needs of EMA or other European regulatory authorities, or to generate the health economics and outcomes research data needed to support pricing and reimbursement negotiations or decisions in Europe, we may have difficulties obtaining marketing authorization for our products from EMA/European Commission and may have difficulties obtaining pricing and reimbursement approval for our products at a national level. Any factors preventing or limiting the market acceptance of our products could have a material adverse effect on our business, results of operations and financial condition.
All of our patents in the Anti-TNF and Fibrosis program are method of use patents, which may result in biosimilar drugs being used without our permission.
The success of our most advanced drug development platform depends on the enforceability of our method of use patents, as there are currently many biosimilar anti-TNF drugs in the market. If we are unable to obtain composition of matter patents, and enforce such patents, our ability to generate revenue from the anti-TNF platform may be significantly limited and competitors may be able to use our research to bring competing drugs to market which would reduce our market share.
The majority of our license agreements provide the licensors and/or counter-parties the right to use and/or exploit such licensed intellectual property.
The majority of our license agreements provide the licensors and/or counter-parties the right to use and/or exploit such licensed intellectual property, and in some cases provide them ownership of such intellectual property, know-how and research results. As such, we may be in competition with parties who we have license agreements with, will likely not have the sole right to monetize, sell or distribute our product candidates and may be subject to restrictions on use and territory of sales. Any or all of the above may have a material adverse effect on our results of operations and cash flows and ultimately the value of our securities.
Interim, topline and preliminary data from our clinical trials may change as more patient data become available, and are subject to audit and verification procedures that could result in material changes in the final data.
From time to time, we may publicly disclose preliminary, interim or topline data from our preclinical studies and clinical trials, which is based on a preliminary analysis of then-available data, and the results and related findings and conclusions are subject to change as patient enrollment and treatment continues and more patient data become available. For example, any positive results from our preclinical testing, Phase 1 and Phase 2 clinical trials of our product candidate for any product candidate may not necessarily be predictive of the results from planned or future clinical trials for such product candidates. Many companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in clinical trials after achieving positive results in pre-clinical and early clinical development, and we cannot be certain that we will not face similar setbacks. These setbacks have been caused by, among other things, pre-clinical findings while clinical trials were underway or safety or efficacy observations in clinical trials, including adverse events. Adverse differences between previous preliminary or interim data and future interim or final data could significantly harm our business prospects. We may also announce topline data following the completion of a preclinical study or clinical trial, which may be subject to change following a more comprehensive review of the data related to the particular study or trial. We also make assumptions, estimations, calculations and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully and carefully evaluate all data. As a result, the interim, topline or preliminary results that we report may differ from future results of the same studies, or different conclusions or considerations may qualify such results, once additional data has been received and fully evaluated. Preliminary, interim, or topline data also remains subject to audit and verification procedures that may result in the final data being materially different from the data we previously published. As a result, preliminary, interim, and topline data should be viewed with caution until the final data is available.
56
Further, others, including regulatory agencies, may not accept or agree with our assumptions, estimates, calculations, conclusions or analyses or may interpret or weigh the importance of data differently, which could impact the value of the particular program, the approvability or commercialization of the particular product candidate or product and our company in general. In addition, the information we choose to publicly disclose regarding a particular study or clinical trial is based on what is typically extensive information, and you or others may not agree with what we determine to be material or otherwise appropriate information to include in our disclosure. Moreover, our interpretation of clinical data or our conclusions based on the preclinical in vitro and in vivo models may prove inaccurate, as preclinical and clinical data can be susceptible to varying interpretations and analyses, and many companies that believed their product candidates performed satisfactorily in preclinical studies and clinical trials nonetheless failed to obtain FDA approval or a marketing authorization granted by the European Commission.
If we fail to produce positive results in our future clinical trials, the development timeline and regulatory approval and commercialization prospects for such product candidates, and, correspondingly, our business and financial prospects, would be materially adversely affected.
We have limited marketing experience, and we may not be able to successfully commercialize any of our future product candidates, even if they are approved in the future.
Our ability to generate revenues ultimately will depend on our ability to sell our approved products and secure adequate third-party reimbursement. We currently have no experience in marketing and selling our products. The commercial success of our future products depends on a number of factors beyond our control, including the willingness of physicians to prescribe our future products to patients, payors’ willingness and ability to pay for our future products, the level of pricing achieved, patients’ response to our future products, and the ability of our future marketing partners to generate sales. There can be no guarantee that we will be able to establish or maintain the personnel, systems, arrangements and capabilities necessary to successfully commercialize our future products or any product candidate approved by the FDA or other regulatory authority in the future. If we fail to establish or maintain successful marketing, sales and reimbursement capabilities or fail to enter into successful marketing arrangements with third parties, our product revenues may suffer.
If the price for any of our future approved products decreases or if governmental and other third-party payors do not provide coverage and adequate reimbursement levels, our revenue and prospects for profitability will suffer.
Patients who are prescribed medicine for the treatment of their conditions generally rely on third-party payors to reimburse all or part of the costs associated with their prescription drugs. Reimbursement systems in international markets vary significantly by country and by region, and reimbursement approvals generally must be obtained on a country-by-country basis. Coverage and adequate reimbursement from governmental healthcare programs, such as Medicare and Medicaid, and commercial payors is critical to new product acceptance. Coverage decisions may depend upon clinical and economic standards that disfavor new drug products when more established or lower cost therapeutic alternatives are already available or subsequently become available. Even if we obtain coverage for our future product candidates, the resulting reimbursement payment rates may require co-payments that patients find unacceptably high. Patients may not use our future product candidates if coverage is not provided or reimbursement is inadequate to cover a significant portion of a patient’s cost.
In addition, the market for our future product candidates in the U.S. will depend significantly on access to third-party payors’ drug formularies, or lists of medications for which third-party payors provide coverage and reimbursement. The industry competition to be included in such formularies often leads to downward pricing pressures on pharmaceutical companies. Also, third-party payors may refuse to include a particular branded drug in their formularies or otherwise restrict patient access to a branded drug when a less costly generic equivalent or other alternative is available.
57
Third-party payors, whether foreign or domestic, or governmental or commercial, are developing increasingly sophisticated methods of controlling healthcare costs. The current environment is putting pressure on companies to price products below what they may feel is appropriate. Our future revenues and overall success could be negatively impacted if we sell future product candidates at less than an optimized price. In addition, in the U.S., no uniform policy of coverage and reimbursement for drug products exists among third-party payors. Therefore, coverage and reimbursement for our future product candidates may differ significantly from payor to payor. As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of our future product candidates to each payor separately, with no assurance that coverage will be obtained. If we are unable to obtain coverage of, and adequate payment levels for, our future product candidates, physicians may limit how much or under what circumstances they will prescribe or administer them and patients may decline to purchase them. This could affect our ability to successfully commercialize our product candidates, and thereby adversely impact our profitability, results of operations, financial condition and future success.
In addition, where we have chosen to collaborate with a third party on product candidate development and commercialization, our partner may elect to reduce the price of our products to increase the likelihood of obtaining reimbursement approvals. In many countries, products cannot be commercially launched until reimbursement is approved and the negotiation process in some countries can exceed 12 months. In addition, pricing and reimbursement decisions in certain countries can be affected by decisions made in other countries, which can lead to mandatory price reductions and/or additional reimbursement restrictions across a number of other countries, which may adversely affect sales and profitability. In the event that countries impose prices that are not sufficient to allow us or our partners to generate a profit, our partners may refuse to launch the product in such countries or withdraw the product from the market, which would adversely affect sales and profitability.
Business interruptions could delay us in the process of developing our future product candidates and could disrupt our product sales.
Loss of our future manufacturing facilities, stored inventory or laboratory facilities through fire, theft or other causes, could have an adverse effect on our ability to meet demand for our future product candidates or to continue product development activities and to conduct our business. Failure to supply our partners with commercial products may lead to adverse consequences, including the right of partners to assume responsibility for product supply. Even if we obtain insurance coverage to compensate us for such business interruptions, such coverage may prove insufficient to fully compensate us for the damage to our business resulting from any significant property or casualty loss to our inventory.
If product liability lawsuits are successfully brought against us, we will incur substantial liabilities and may be required to limit the commercialization of our future product candidates.
Although we have never had any product liability claims or lawsuits brought against us, we face potential product liability exposure related to the testing of our future product candidates in human clinical trials, and we will face exposure to claims in jurisdictions where we market and distribute in the future. We may face exposure to claims by an even greater number of persons when we begin marketing and distributing our products commercially in the U.S. and elsewhere. In the future, an individual may bring a liability claim against us alleging that one of our future product candidates caused an injury. While we continue to take what we believe are appropriate precautions, we may be unable to avoid significant liability if any product liability lawsuit is brought against us. Large judgments have been awarded in class action or individual lawsuits based on drugs that had unanticipated side effects. Although we have purchased insurance to cover product liability lawsuits, if we cannot successfully defend our company against product liability claims, or if such insurance coverage is inadequate, we will incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in decreased demand for our products, reputational damage, withdrawal of clinical trial participation participants, litigation costs, product recall costs, monetary awards, increased costs for liability insurance, lost revenues and business interruption.
58
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and legal requirements.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to comply with FDA, SEC or Office of Inspector General regulations, or regulations of any other applicable regulatory authority, failure to provide accurate information to the FDA or the SEC, comply with applicable manufacturing standards, other federal, state or foreign laws and regulations, report information or data accurately or disclose unauthorized activities. Employee misconduct could also involve the improper use of information, including information obtained in the course of clinical trials, or illegal appropriation of drug product, which could result in government investigations and serious harm to our reputation. Despite our adoption of a Code of Ethics, employee misconduct is not always possible to identify and deter. The precautions we take to detect and prevent these prohibited activities may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against our company, and we are not successful in defending our company or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
We are subject to the U.S. Foreign Corrupt Practices Act and other anti-corruption laws, as well as export control laws, customs laws, sanctions laws and other laws governing our operations. If we fail to comply with these laws, we could be subject to civil or criminal penalties, other remedial measures, and legal expenses, which could adversely affect our business, results of operations and financial condition.
Our operations are subject to anti-corruption laws, including the U.S. Foreign Corrupt Practices Act (“FCPA”), and other anti-corruption laws that apply in countries in which we do business. The FCPA and these other laws generally prohibit our company and our employees and intermediaries from bribing, being bribed or making other prohibited payments to government officials or other persons to obtain or retain business or gain some other business advantage. We and our commercial partners operate in a number of jurisdictions that pose a high risk of potential FCPA violations, and we participate in collaborations and relationships with third parties whose actions could potentially subject us to liability under the FCPA or local anti-corruption laws. In addition, we cannot predict the nature, scope or effect of future regulatory requirements to which our international operations might be subject or the manner in which existing laws might be administered or interpreted.
We are also subject to other laws and regulations governing our international operations, including regulations administered by the governments of the U.S., Canada, Israel, the United Kingdom and authorities in the EU, including applicable export control regulations, economic sanctions on countries and persons, customs requirements and currency exchange regulations, collectively referred to as the Trade Control Laws.
However, there is no assurance that we will be completely effective in ensuring our compliance with all applicable anti-corruption laws, including the FCPA or other legal requirements, including Trade Control Laws. If we are not in compliance with the FCPA and other anti-corruption laws or Trade Control Laws, we may be subject to criminal and civil penalties, disgorgement and other sanctions and remedial measures, and legal expenses, which could have an adverse impact on our business, financial condition, results of operations and liquidity. Likewise, any investigation of any potential violations of the FCPA, other anti-corruption laws by the U.S. or other authorities could also have an adverse impact on our reputation, business, financial condition and results of operations.
Security breaches, loss of data and other disruptions could compromise sensitive information related to our business, prevent us from accessing critical information or expose us to liability, which could adversely affect our business and our reputation.
In the ordinary course of business, we expect to collect and store sensitive data, including legally protected patient health information, credit card information, personally identifiable information about our employees, intellectual property, and proprietary business information. We expect to manage and maintain our applications and data utilizing on-site systems. These applications and data encompass a wide variety of business-critical information including research and development information, commercial information and business and financial information.
59
The secure processing, storage, maintenance and transmission of this critical information is vital to our operations and business strategy, and we devote significant resources to protecting such information. Although we take measures to protect sensitive information from unauthorized access or disclosure, our information technology and infrastructure may be vulnerable to attacks by hackers, or viruses, breaches or interruptions due to employee error, malfeasance or other disruptions, or lapses in compliance with privacy and security mandates. Any such virus, breach or interruption could compromise our networks and the information stored there could be accessed by unauthorized parties, publicly disclosed, lost or stolen. We have measures in place that are designed to prevent, and if necessary, to detect and respond to such security incidents and breaches of privacy and security mandates. However, in the future, any such access, disclosure or other loss of information could result in legal claims or proceedings, liability under laws that protect the privacy of personal information, such as HIPAA and European Union General Data Protection Regulation (“GDPR”), government enforcement actions and regulatory penalties. Unauthorized access, loss or dissemination could also disrupt our operations, including our ability to process samples, provide test results, share and monitor safety data, bill payors or patients, provide customer support services, conduct research and development activities, process and prepare company financial information, manage various general and administrative aspects of our business and may damage our reputation, any of which could adversely affect our business, financial condition and results of operations.
In May 2016, the EU formally adopted the GDPR, which applies to all EU member states and became effective on May 25, 2018 and replaced the European Union Data Protection Directive. The regulation introduces stringent new data protection requirements in the EU and substantial fines for breaches of the data protection rules. It may increase our responsibility and liability in relation to personal data that we process and we may be required to put in place additional mechanisms ensuring compliance with the new data protection rules. The GDPR is a complex law and the regulatory guidance is still evolving, including with respect to how the GDPR should be applied in the context of clinical trials or other transactions from which we may gain access to personal data. GDPR increases our costs of compliance and results in greater legal risks.
Our research and development programs and product candidates are in development. As a result, we are unable to predict if or when we will successfully develop or commercialize our product candidates.
Our clinical-stage product candidates as well as our other drug pipeline candidates will require significant further investment and regulatory approvals prior to commercialization. Each of our product candidates will require the selection of suitable patients for our clinical trials and additional clinical development, management of clinical, preclinical and manufacturing activities, obtaining regulatory approval, obtaining manufacturing supply, building of a commercial organization, substantial investment and significant marketing efforts before we generate any revenues from product sales. We are not permitted to market or promote any of our product candidates before we receive regulatory approval from the FDA or comparable foreign regulatory authorities, and we may never receive such regulatory approval for any of our product candidates. By such time, if ever, as we may receive necessary regulatory approvals for our product candidates, the standard of care for the treatment of diseases associated with our product candidates may have evolved such that it would be necessary to modify our plans for full approval and commercial acceptance of our products may be limited by a change in the standard of care.
Even if we obtain the required financing or establish a collaboration to enable us to conduct late-stage clinical development of our product candidates and pipeline assets, we cannot be certain that such clinical development would be successful, or that we will obtain regulatory approval or be able to successfully commercialize any of our product candidates and generate revenue. Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful, and the clinical trial process may fail to demonstrate that our product candidates are safe and effective for their proposed uses. Any such failure could cause us to abandon further development of any one or more of our product candidates and may delay development of other product candidates. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy traits despite having progressed through preclinical studies and initial clinical trials. Any delay in, or termination of, our clinical trials will delay and possibly preclude the submission of any NDAs with the FDA and, ultimately, our ability to commercialize our product candidates and generate product revenue.
60
We have not previously submitted an NDA to the FDA, or similar drug approval filings to comparable foreign authorities, for any product candidate, and we cannot be certain that any of our product candidates will receive regulatory approval. Further, our product candidates may not receive regulatory approval even if they are successful in clinical trials. If we do not receive regulatory approvals for our product candidates, we may not be able to continue our operations. Even if we successfully obtain regulatory approvals to market one or more of our product candidates, our revenues will be dependent, in part, upon our or our collaborators’ and future collaborators’ ability to obtain regulatory approval for the companion diagnostics to be used with our product candidates, if required, and upon the size of the markets in the territories for which we gain regulatory approval and have commercial rights. If the markets for patient subsets that we are targeting are not as significant as we estimate, we may not generate significant revenues from sales of such products, if approved.
Further, even if any product candidate we develop was to receive marketing approval or be commercialized for use in combination with other existing therapies, we would continue to bear the risks that the FDA or similar foreign regulatory authorities could revoke approval of the therapy used in combination with our product candidate or that safety, efficacy, manufacturing or supply issues could arise with these existing therapies.
The successful commercialization of our product candidates, if approved, will depend on achieving market acceptance and we may not be able to gain sufficient acceptance to generate significant revenue.
Even if our product candidates are successfully developed and receive regulatory approval, they may not gain market acceptance among physicians, patients, healthcare payors such as private insurers or governments and other funding parties and the medical community. The degree of market acceptance for any of our products will depend on a number of factors, including:
| ● | demonstration of the clinical efficacy and safety of our products; |
| ● | the prevalence and severity of any adverse side effects; |
| ● | limitations or warnings contained in the product’s approved labeling; |
| ● | cost-effectiveness and availability of acceptable pricing; |
| ● | competitive product profile versus alternative treatment methods and the superiority of alternative treatment or therapeutics; |
| ● | the effectiveness of marketing and distribution methods and support for the products; and |
| ● | the availability of coverage and adequate reimbursement from third-party payors to the extent that our products receive regulatory approval. |
61
Disease indications may be small subsets of a disease that could be parsed into smaller and smaller indications as different subsets of diseases are defined. This increasingly fine characterization of diseases could have negative consequences; including creating an approved indication that is so small as not to have a viable market for us. If future technology allows characterization of a disease in a way that is different from the characterization used for large pivotal studies, it may make those studies invalid or reduce their usefulness, and may require repeating all or a portion of the studies. Future technology may supply better prognostic ability which could reduce the portion of patients projected to need a new therapy. Even after being cleared by regulatory authorities, a product may later be shown to be unsafe or not to have its purported effect, thereby preventing its widespread use or requiring withdrawal from the market.
We may not be successful in establishing development and commercialization collaborations which could adversely affect, and potentially prohibit, our ability to develop our product candidates.
Developing pharmaceutical products, conducting clinical trials, obtaining regulatory approval, establishing manufacturing capabilities and marketing approved products is expensive, and therefore we may seek to enter into additional collaborations with companies that have more resources and experience in order to continue to develop and commercialize our product candidates. We also may be required due to financial or scientific constraints to enter into additional collaboration agreements to research and/or to develop and commercialize our product candidates. The establishment and realization of such collaborations may not be possible or may be problematic. There can be no assurance that we will be able to establish such additional collaborations on favorable terms, if at all, or that our current or future collaborative arrangements will be successful or maintained for any specific product candidate or indication. If we are unable to reach successful agreements with suitable collaboration partners for the ongoing development and commercialization of our product candidates, we may face increased costs, we may be forced to limit the scope and number of our product candidates we can commercially develop or the territories in which we commercialize such product candidates, and we may be unable to commercialize products or programs for which a suitable collaboration partner cannot be found. If we fail to achieve successful collaborations, our operating results and financial condition will be materially and adversely affected.
In addition, the terms of any collaboration agreements may place restrictions on our activities with respect to other products, including by limiting our ability to grant licenses or develop products with other third parties, or in different indications, diseases or geographical locations, or may place additional obligations on us with respect to development or commercialization of our product candidates. If we fail to comply with or breach any provision of a collaboration agreement, a collaborator may have the right to terminate, in whole or in part, such agreement or to seek damages.
Some of our collaboration and licensing agreements are complex and involve sharing or division of ownership of certain data, know-how and intellectual property rights among the various parties. Accordingly, our collaborators could interpret certain provisions differently than we or our other collaborators which could lead to unexpected or inadvertent disputes with collaborators. In addition, these agreements might make additional collaborations, partnering or mergers and acquisitions difficult.
There is no assurance that a collaborator who is acquired by a third party would not attempt to change certain contract provisions that could negatively affect our collaboration. The acquiring company may also not accept the terms or assignment of our contracts and may seek to terminate the agreements. Any one of our collaborators could breach covenants, restrictions and/or sub-license agreement provisions leading us into disputes and potential breaches of our agreements with other partners.
We will not be able to successfully commercialize our product candidates without establishing sales and marketing capabilities internally or through collaborators.
We may not be able to find suitable sales and marketing staff and collaborators for all of our product candidates. The development of a marketing and sales capability will require significant expenditures, management resources and time. The cost of establishing such a sales force may exceed any potential product revenue, or our marketing and sales efforts may be unsuccessful. If we are unable to develop an internal marketing and sales capability in a timely fashion, or at all, or if we are unable to enter into a marketing and sales arrangement with a third party on acceptable terms, we may be unable to effectively market and sell approved products, if any, which would prevent us from being able to generate revenue and attain profitability. Further, we may not develop an internal marketing and sales capability if we are unable to successfully develop and seek regulatory approval for our product candidates.
62
We will rely upon third-party contractors and service providers for the execution of some aspects of our development programs. Failure of these collaborators to provide services of a suitable quality and within acceptable timeframes may cause the delay or failure of our development programs.
We outsource certain functions, tests and services to third parties, partners, medical institutions and collaborators and plan to outsource manufacturing to collaborators and/or contract manufacturers, and we will rely on third parties for quality assurance, clinical monitoring, clinical data management and regulatory expertise. In particular, we rely on our partners to run our clinical trials. There is no assurance that such individuals or organizations will be able to provide the functions, tests, drug supply or services as agreed upon or to acceptable quality standards, and we could suffer significant delays in the development of our products or processes. In particular, certain third party service providers may be unable to comply with their contractual obligations to us due to disruptions caused by the COVID-19 pandemic, including reduced operations or headcount reductions, or otherwise, and in certain cases we may have limited recourse if the non-compliance is due to factors outside of the service provider’s control.
The timelines of our clinical trials may be impacted by numerous factors and any delays may adversely affect our ability to execute our current business strategy.
Clinical testing is expensive, difficult to design and implement, can take many years to complete, and is uncertain as to outcome. We may experience delays in clinical trials at any stage of development and testing of our product candidates. Our planned clinical trials may not begin on time, have an effective design, enroll a sufficient number of subjects, or be completed on schedule, if at all.
Events which may result in a delay or unsuccessful completion of clinical trials include:
| ● | inability to raise funding necessary to initiate or continue a trial; |
| ● | delays in obtaining regulatory approval to commence a trial; |
| ● | delays in reaching agreement with the FDA on final trial design; |
| ● | imposition of a clinical hold following an inspection of our clinical trial operations or trial sites by the FDA or other regulatory authorities; |
| ● | delays in reaching agreement on acceptable terms with prospective CROs and clinical trial sites; |
| ● | delays in obtaining required institutional review board approval at each site; |
| ● | delays in having subjects complete participation in a trial or return for post-treatment follow-up; |
| ● | delays caused by subjects dropping out of a trial due to side effects or otherwise; |
| ● | clinical sites dropping out of a trial to the detriment of enrollment; |
| ● | time required to add new clinical sites; and |
| ● | delays by our contract manufacturers to produce and deliver a sufficient supply of clinical trial materials. |
63
If initiation or completion of any of our clinical trials for our product candidates are delayed for any of the above reasons or for other reasons, our development costs may increase, our approval process could be delayed, any periods after commercial launch and before expiration of patent protection may be reduced and our competitors may have more time to bring products to market before we do. Any of these events could impair the commercial potential of our product candidates and could have a material adverse effect on our business.
Clinical trials of our product candidates may not uncover all possible adverse effects that patients may experience.
Clinical trials are conducted in representative samples of the potential patient population, which may have significant variability. Clinical trials are by design based on a limited number of subjects and of limited duration for exposure to the product used to determine whether, on a potentially statistically significant basis, the planned safety and efficacy of any product candidate can be achieved. As with the results of any statistical sampling, we cannot be sure that all side effects of our product candidates may be uncovered, and it may be the case that only with a significantly larger number of patients exposed to the product candidate for a longer duration, that a more complete safety profile is identified. Further, even larger clinical trials may not identify rare serious adverse effects or the duration of such studies may not be sufficient to identify when those events may occur. Patients treated with our products, if approved, may experience adverse reactions and it is possible that the FDA or other regulatory authorities may ask for additional safety data as a condition of, or in connection with, our efforts to obtain approval of our product candidates. If safety problems occur or are identified after our product candidates reach the market, we may, or regulatory authorities may require us to amend the labeling of our products, recall our products or even withdraw approval for our products.
Failure can occur at any stage of our drug development efforts.
We may experience numerous unforeseen events during, or as a result of, testing that could delay or prevent us from obtaining regulatory approval for, or commercializing our drug candidates, including but not limited to:
| ● | regulators or Institutional Review Boards (IRBs) may not authorize us to commence a clinical trial or conduct a clinical trial at a prospective trial site; |
| ● | conditions may be imposed upon us by the FDA regarding the scope or design of our clinical trials, or we may be required to resubmit our clinical trial protocols to IRBs for review due to changes in the regulatory environment; |
| ● | the number of subjects required for our clinical trials may be larger, patient enrollment may take longer, or patients may drop out of our clinical trials at a higher rate than we anticipate; |
| ● | we may have to suspend or terminate one or more of our clinical trials if we, regulators, or IRBs determine that the participants are being subjected to unreasonable health risks; |
| ● | our third-party contractors, clinical investigators or contractual collaborators may fail to comply with regulatory requirements or fail to meet their contractual obligations to us in a timely manner; |
| ● | the FDA may not accept clinical data from trials that are conducted at clinical sites in countries where the standard of care is potentially different from the United States; |
| ● | our tests may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional testing; and |
| ● | the costs of our pre-clinical and/or clinical trials may be greater than we anticipate. |
64
We rely on third parties to conduct our pre-clinical studies and clinical studies and trials, and if they do not perform their obligations to us we may not be able to obtain approval for additional indications.
We do not currently have the ability to independently conduct pre-clinical studies or clinical studies and trials, and we have to date relied on third parties, such as third-party contract research and governmental organizations and medical institutions to conduct studies and trials for us. Our reliance on third parties for development activities reduces our control over these activities. These third parties may not complete activities on schedule or may not conduct our pre-clinical studies and our clinical studies and trials in accordance with regulatory requirements or our study design. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may be adversely affected, and our efforts to obtain regulatory approvals for and commercialize indications may be delayed.
If we conduct studies with other parties, we may not have control over all decisions associated with that trial. To the extent that we disagree with the other party on such issues as study design, study timing and the like, it could adversely affect our drug development plans.
Although we also rely on third parties to manage the data from our studies and trials, we are responsible for confirming that each of our studies and trials is conducted in accordance with its general investigational plan and protocol. Moreover, the FDA and foreign regulatory agencies will require us to comply with applicable regulations and standards, including Good Laboratory Practice (GLP) and Good Clinical Practice (GCP), for conducting, recording and reporting the results of such studies and trials to assure that the data and the results are credible and accurate and that the human study and trial participants are adequately protected. Our reliance on third-parties does not relieve us of these obligations and requirements, and we may fail to obtain regulatory approval for any additional indications if these requirements are not met.
We will need to continue to develop and maintain distribution and production capabilities or relationships to be successful.
We may not be able to successfully manufacture any product, either independently or under manufacturing arrangements, if any, with third party manufacturers. Moreover, if any manufacturer should cease doing business with us or experience delays, shortages of supply or excessive demands on their capacity, we may not be able to obtain adequate quantities of product in a timely manner, or at all. Manufacturers, and in certain situations their suppliers, are required to comply with current NDA commitments and current good manufacturing practices (cGMP) requirements enforced by the FDA, and similar requirements of other countries. The failure by a manufacturer to comply with these requirements could affect its ability to provide us with products. Although we intend to rely on third-party contract manufacturers, we are ultimately responsible for ensuring that our products are manufactured in accordance with cGMP. In addition, if, during a preapproval inspection or other inspection of our third-party manufacturers’ facility or facilities, the FDA determines that the facility is not in compliance with cGMP, any of our marketing applications that lists such facility as a manufacturer may not be approved or approval may be delayed until the facility comes into compliance with cGMP and completes a successful re-inspection by the FDA.
Any manufacturing problem, natural disaster, or epidemic, affecting manufacturing facilities, or the loss of a contract manufacturer could be disruptive to our operations and result in lost sales. Additionally, we will be reliant on third parties to supply the raw materials needed to manufacture our products. Any reliance on suppliers may involve several risks, including a potential inability to obtain critical materials and reduced control over production costs, delivery schedules, reliability and quality. Any unanticipated disruption to future contract manufacture caused by problems at suppliers could delay shipment of products, increase our cost of goods sold and result in lost sales. If our suppliers were to be unable to supply us with adequate supply of our drugs, it could have a material adverse effect on our ability to successfully commercialize our drug candidates.
65
If we rely on a sole source of supply to manufacture our products we could be impacted by the viability of our supplier.
We intend to attempt to source our products from more than one supplier. We also intend to enter into contracts with any supplier of our products to contractually obligate them to meet our requirements. However, if we are reliant on a single supplier and that supplier cannot or will not meet our requirements (for whatever reason), our business could be adversely impacted.
We may not be able to sufficiently scale-up manufacturing of our drug candidates.
We may not be able to successfully increase in a sufficient manner the manufacturing capacity for our drug candidates, whether in collaboration with third-party manufacturers or on our own, in a timely or cost-effective manner or at all. If a contract manufacturer makes improvements in the manufacturing process for our drug candidates, we may not own, or may have to share, the intellectual property rights to those improvements.
Significant scale-up of manufacturing may require additional validation studies, which are costly and which the FDA must review and approve. In addition, quality issues may arise during those scale-up activities because of the inherent properties of a drug candidate itself or of a drug candidate in combination with other components added during the manufacturing and packaging process, or during shipping and storage of the finished product or active pharmaceutical ingredients. If we are unable to successfully scale-up manufacture of any of our drug candidates in sufficient quality and quantity, the development of that drug candidate and regulatory approval or commercial launch for any resulting drug products may be delayed or there may be a shortage in supply, which could significantly harm our business.
Risks Related to Development and Regulatory Approval of our Future Product Candidates
Clinical trials are expensive, time-consuming, uncertain and susceptible to change, delay or termination. The results of clinical trials are open to differing interpretations.
We have three separate programs for producing anti-inflammatory agents: (1) investigating new clinical opportunities for anti-TNF, (2) identifying orally available, small molecules that are agonists of α7 nicotinic acetylcholine receptor, and (3) identifying patentable analogs of CBD that initially will be used as pain medications. However, these programs, including the related clinical trials, are expensive, time consuming and difficult to design and implement.
Regulatory agencies may analyze or interpret the results of clinical trials differently than us. Even if the results of our clinical trials are favorable, the clinical trials for a number of its future product candidates are expected to continue for several years and may take significantly longer to complete. In addition, the FDA or other regulatory authorities, including state, local and foreign authorities, or an IRB, with respect to a trial at our institution, may suspend, delay or terminate its clinical trials at any time, require us to conduct additional clinical trials, require a particular clinical trial to continue for a longer duration than originally planned, require a change to its development plans such that we conduct clinical trials for a product candidate in a different order, e.g., in a step-wise fashion rather than running two trials of the same product candidate in parallel, or could suspend or terminate the registrations and quota allotments we require in order to procure and handle controlled substances, for various reasons, including the following, any of which could have a material adverse effect on our business, financial condition and results of operations:
| ● | lack of effectiveness of any product candidate during clinical trials; |
| ● | discovery of serious or unexpected toxicities or side effects experienced by trial participants or other safety issues, such as drug interactions, including those which cause confounding changes to the levels of other concomitant medications; |
| ● | slower than expected rates of subject recruitment and enrollment rates in clinical trials; |
66
| ● | difficulty in retaining subjects who have initiated a clinical trial but may withdraw at any time due to adverse side effects from the therapy, insufficient efficacy, fatigue with the clinical trial process or for any other reason; |
| ● | delays or inability in manufacturing or obtaining sufficient quantities of materials for use in clinical trials due to regulatory and manufacturing constraints; |
| ● | inadequacy of or changes in our manufacturing process or product formulation; |
| ● | delays in obtaining regulatory authorization to commence a trial, including “clinical holds” or delays requiring suspension or termination of a trial by a regulatory agency, such as the FDA, before or after a trial is commenced; |
| ● | DEA related recordkeeping, reporting security or other violations at a clinical site, leading the DEA or state authorities to suspend or revoke the site’s-controlled substance license and causing a delay or termination of planned or ongoing trials; |
| ● | changes in applicable regulatory policies and regulation, including changes to requirements imposed on the extent, nature or timing of studies; |
| ● | delays or failure in reaching agreement on acceptable terms in clinical trial contracts or protocols with prospective clinical trial sites; |
| ● | uncertainty regarding proper dosing; |
| ● | delay or failure to supply product for use in clinical trials which conforms to regulatory specification; |
| ● | unfavorable results from ongoing pre-clinical studies and clinical trials; |
| ● | failure of our contract research organizations (“CROs”), or other third-party contractors to comply with all contractual requirements or to perform their services in a timely or acceptable manner; |
| ● | failure by our company, our employees, our CROs or their employees to comply with all applicable FDA or other regulatory requirements relating to the conduct of clinical trials; |
| ● | scheduling conflicts with participating clinicians and clinical institutions; |
| ● | failure to design appropriate clinical trial protocols; |
| ● | regulatory concerns with CBD derivative products generally and the potential for abuse, despite only working with non-plant based non-psychoactive products; |
| ● | insufficient data to support regulatory approval; |
| ● | inability or unwillingness of medical investigators to follow our clinical protocols; or |
| ● | difficulty in maintaining contact with patients during or after treatment, which may result in incomplete data. |
We may find it difficult to enroll patients in our clinical trials, which could delay or prevent clinical trials of our product candidates.
Identifying and qualifying patients to participate in clinical trials of our product candidates is critical to our success. The timing of our clinical trials depends on the speed at which we can recruit eligible patients to participate in testing our product candidates. We have experienced delays in some of our clinical trials, and we may experience similar delays in the future. These delays could result in increased costs, delays in advancing our product development, or termination of the clinical trials altogether.
67
We may not be able to identify, recruit and enroll a sufficient number of patients, or those with required or desired characteristics to achieve diversity in a study, to complete our clinical trials within the expected timeframe. Patient enrollment can be impacted by factors including, but not limited to:
| ● | design and complexity and/or commitment of participation required in the study protocol; | |
| ● | size of the patient population; |
| ● | eligibility criteria for the study in question; | |
| ● | clinical supply availability; |
| ● | delays in participating site identification, qualification and subsequent activation to enroll; | |
| ● | perceived risks and benefits of the product candidate under study, including as a result of adverse effects observed in similar or competing therapies; |
| ● | proximity and availability of clinical trial sites for prospective patients; | |
| ● | availability of competing therapies and clinical trials; |
| ● | competition of site efforts to facilitate timely enrollment in clinical trials; | |
| ● | participating site motivation; |
| ● | patient referral practices of physicians; | |
| ● | activities of patient advocacy groups; |
| ● | ability to monitor patients adequately during and after treatment; and | |
| ● | severity of the disease under investigation. |
In particular, each of the conditions for which we plan to evaluate our product candidates are diseases with limited patient pools from which to draw for clinical trials. The eligibility criteria of our clinical trials will further limit the pool of available study participants. Additionally, the process of finding and diagnosing patients may prove costly. The treating physicians in our clinical trials may also use their medical discretion in advising patients enrolled in our clinical trials to withdraw from our studies to try alternative therapies. In addition, the COVID-19 pandemic may impact patient ability and willingness to travel to clinical trial sites as a result of quarantines and other restrictions, which may negatively impact enrollment in our clinical trials.
We may not be able to initiate or continue clinical trials if we cannot enroll the required eligible patients per protocol to participate in the clinical trials required by the FDA or the EMA or other regulatory agencies. Our ability to successfully initiate, enroll and complete a clinical trial in any foreign country is subject to numerous risks unique to conducting business in foreign countries, including:
| ● | difficulty in establishing or managing relationships with contract research organizations (“CROs”) and physicians; | |
| ● | different standards for the conduct of clinical trials; |
| ● | our inability to locate qualified local consultants, physicians and partners; | |
| ● | the potential burden of complying with a variety of foreign laws, medical standards and regulatory requirements, including the regulation of pharmaceutical and biotechnology products and treatment; |
| ● | ability to procure and deliver necessary clinical trial materials needed to perform the study; and | |
| ● | inability to implement adequate training at participating sites remotely when in person training cannot be completed. |
68
If we have difficulty enrolling a sufficient number of patients to conduct our clinical trials as planned, we may need to delay, limit or terminate ongoing or planned clinical trials, any of which would have an adverse effect on our ability to complete clinical trials and ultimately our results of operations.
Any failure by our company to comply with existing regulations could harm our reputation and operating results.
We are subject to extensive regulation by U.S. federal and state and foreign governments in each of the U.S., European and Canadian markets, in which we plan to sell our products, or in markets where we have product candidates progressing through the approval process.
We must adhere to all regulatory requirements including FDA’s Good Laboratory Practice (“GLP”), Good Clinical Practice (“GCP”), and GMP requirements, pharmacovigilance requirements, advertising and promotion restrictions, reporting and recordkeeping requirements, and their European equivalents. If we or our suppliers fail to comply with applicable regulations, including FDA pre-or post-approval requirements, then the FDA or other foreign regulatory authorities could sanction our company. Even if a drug is approved by the FDA or other competent authorities, regulatory authorities may impose significant restrictions on a product’s indicated uses or marketing or impose ongoing requirements for potentially costly post-marketing trials. Any of product candidates which may be approved in the U.S. will be subject to ongoing regulatory requirements for manufacturing, labeling, packaging, storage, distribution, import, export, advertising, promotion, sampling, recordkeeping and submission of safety and other post-market information, including both federal and state requirements. In addition, manufacturers and manufacturers’ facilities are required to comply with extensive FDA requirements, including ensuring that quality control and manufacturing procedures conform to GMP. As such, we and our contract manufacturers (in the event contract manufacturers are appointed in the future) are subject to continual review and periodic inspections to assess compliance with GMP. Accordingly, we and others with whom we work will have to spend time, money and effort in all areas of regulatory compliance, including manufacturing, production, quality control and quality assurance. We will also be required to report certain adverse reactions and production problems, if any, to the FDA, and to comply with requirements concerning advertising and promotion for its products. Promotional communications with respect to prescription drugs are subject to a variety of legal and regulatory restrictions and must be consistent with the information in the product’s approved label. Similar restrictions and requirements exist in the EU and other markets where we operate.
If a regulatory agency discovers previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, or disagrees with the promotion, marketing or labeling of the product, it may impose restrictions on that product or on our company, including requiring withdrawal of the product from the market. If we fail to comply with applicable regulatory requirements, a regulatory agency or enforcement authority may issue warning letters, impose civil or criminal penalties, suspend regulatory approval, suspend any of our ongoing clinical trials, refuse to approve pending applications or supplements to approved applications submitted by us, impose restrictions on our operations, or seize or detain products or require a product recall.
In addition, it is possible that our future products will be regulated by the DEA, under the Controlled Substances Act or under similar laws elsewhere. DEA scheduling is a separate process that can delay when a drug may become available to patients beyond an NDA approval date, and the timing and outcome of such DEA process is uncertain. See also “Risks Related to Controlled Substances.”
In addition, any government investigation of alleged violations of law could require us to spend significant time and resources in response, and could generate negative publicity. Any failure to comply with ongoing regulatory requirements may significantly and adversely affect our ability to commercialize and generate revenue from our future product candidates. If regulatory sanctions are applied or if regulatory approval is withdrawn, the value of our business and our operating results may be adversely affected.
69
Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses, divert our management’s attention from the operation of our business and damage our reputation. We expect to spend significant resources on compliance efforts and such expenses are unpredictable. Changing laws, regulations and standards might also create uncertainty, higher expenses and increase insurance costs. As a result, we intend to invest all reasonably necessary resources to comply with evolving standards, and this investment might result in increased management and administrative expenses and a diversion of management time and attention from revenue-generating activities to compliance activities.
We are subject to federal, state and foreign healthcare laws and regulations and implementation of or changes to such healthcare laws and regulations could adversely affect our business and results of operations.
In both the U.S. and certain foreign jurisdictions, there have been a number of legislative and regulatory proposals to change the healthcare system in ways that could impact our ability to sell our future product candidates. If we are found to be in violation of any of these laws or any other federal, state or foreign regulations, we may be subject to administrative, civil and/or criminal penalties, damages, fines, individual imprisonment, we from federal health care programs and the restructuring of our operations. Any of these could have a material adverse effect on our business and financial results. Since many of these laws have not been fully interpreted by the courts, there is an increased risk that we may be found in violation of one or more of their provisions. Any action against us for violation of these laws, even if we ultimately are successful in our defense, will cause us to incur significant legal expenses and divert our management’s attention away from the operation of our business. In addition, in many foreign countries, particularly the countries of the EU the pricing of prescription drugs is subject to government control.
In some foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country.
For example, some EU jurisdictions operate positive and negative list systems under which products may only be marketed once a reimbursement price has been agreed. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical trials that compare the cost-effectiveness of a particular product candidate to currently available therapies. Other member states allow companies to fix their own prices for medicines but monitor and control company profits. Such differences in national pricing regimes may create price differentials between EU member states. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our products. Historically, products launched in the U.K. and EU do not follow price structures of the U.S. In the U.K. and EU, the downward pressure on healthcare costs in general, particularly prescription medicines, has become intense. As a result, barriers to entry of new products are becoming increasingly high and patients are unlikely to use a drug product that is not reimbursed by their government. We may face competition from lower-priced products in foreign countries that have placed price controls on pharmaceutical products. In addition, the importation of foreign products may compete with any future product that we may market, which could negatively impact our profitability.
Specifically, in the U.S., we expect that the 2010 Affordable Care Act (“ACA”), as well as other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we may receive for any approved product. There have been judicial challenges to certain aspects of the ACA and numerous legislative attempts to repeal and/or replace the ACA in whole or in part, and we expect there will be additional challenges and amendments to the ACA in the future. At this time, the full effect that the ACA will have on our business in the future remains unclear. An expansion in the government’s role in the U.S. healthcare industry may cause general downward pressure on the prices of prescription drug products, lower reimbursements or any other product for which we obtain regulatory approval, reduce product utilization and adversely affect our business and results of operations. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability, or commercialize any of our future product candidates for which we may receive regulatory approval.
70
Information obtained from expanded access studies may not reliably predict the efficacy of our future product candidates in company-sponsored clinical trials and may lead to adverse events that could limit approval.
The expanded access studies we are currently supporting are uncontrolled, carried out by individual investigators and not typically conducted in strict compliance with GCPs, all of which can lead to a treatment effect which may differ from that in placebo-controlled trials. These studies provide only anecdotal evidence of efficacy for regulatory review. These studies contain no control or comparator group for reference and this patient data is not designed to be aggregated or reported as study results. Moreover, data from such small numbers of patients may be highly variable. Information obtained from these studies, including the statistical principles that we and the independent investigators have chosen to apply to the data, may not reliably predict data collected via systematic evaluation of the efficacy in company-sponsored clinical trials or evaluated via other statistical principles that may be applied in those trials. Reliance on such information to design our clinical trials may lead to trials that are not adequately designed to demonstrate efficacy and could delay or prevent our ability to seek approval of our future product candidates.
Expanded access programs provide supportive safety information for regulatory review. Physicians conducting these studies may use our future product candidates in a manner inconsistent with the protocol, including in children with conditions beyond those being studied in trials which we sponsor. Any adverse events or reactions experienced by subjects in the expanded access program may be attributed to our future product candidates and may limit our ability to obtain regulatory approval with labeling that we consider desirable, or at all.
There is a high rate of failure for drug candidates proceeding through clinical trials.
Generally, there is a high rate of failure for drug candidates proceeding through clinical trials. We may suffer significant setbacks in our clinical trials similar to the experience of a number of other companies in the pharmaceutical and biotechnology industries, even after receiving promising results in earlier trials. Further, even if we view the results of a clinical trial to be positive, the FDA or other regulatory authorities may disagree with our interpretation of the data. In the event that we obtain negative results from clinical trials for product candidates or other problems related to potential chemistry, manufacturing and control issues or other hurdles occur and our future product candidates are not approved, we may not be able to generate sufficient revenue or obtain financing to continue our operations, our ability to execute on our current business plan may be materially impaired, and our reputation in the industry and in the investment community might be significantly damaged. In addition, our inability to properly design, commence and complete clinical trials may negatively impact the timing and results of our clinical trials and ability to seek approvals for our drug candidates.
If we are found in violation of federal or state “fraud and abuse” laws or similar laws in other jurisdictions, we may be required to pay a penalty and/or be suspended from participation in federal or state health care programs, which may adversely affect our business, financial condition and results of operations.
In the U.S., we are subject to various federal and state health care “fraud and abuse” laws, including anti-kickback laws, false claims laws and other laws intended to reduce fraud and abuse in federal and state health care programs, which could affect our company particularly upon successful commercialization of our products in the U.S. The Medicare and Medicaid Patient Protection Act of 1987, or federal Anti-Kickback Statute, makes it illegal for any person, including a prescription drug manufacturer (or a party acting on its behalf), to knowingly and willfully solicit, receive, offer or pay any remuneration that is intended to induce the referral of business, including the purchase, order or prescription of a particular drug for which payment may be made under a federal health care program, such as Medicare or Medicaid. Under federal law, some arrangements, known as safe harbors, are deemed not to violate the federal Anti-Kickback Statute. Although we seek to structure our business arrangements in compliance with all applicable requirements, it is often difficult to determine precisely how the law will be applied in specific circumstances. Accordingly, it is possible that our practices may be challenged under the federal Anti-Kickback Statute and Federal False Claims Act. Violations of fraud and abuse laws may be punishable by criminal and/or civil sanctions, including fines and/or exclusion or suspension from federal and state health care programs such as Medicare and Medicaid and debarment from contracting with the U.S. government. In addition, private individuals have the ability to bring actions on behalf of the government under the federal False Claims Act as well as under the false claims laws of several states.
71
While we believe that we have structured our business arrangements to comply with these laws, the government could allege violations of, or convict us of violating, these laws. If we are found in violation of one of these laws, we could be required to pay a penalty and could be suspended or excluded from participation in federal or state health care programs, and our business, results of operations and financial condition may be adversely affected.
The Member States of the EU and other countries also have anti-kickback laws and can impose penalties in case of infringement, which, in some jurisdictions, can also be enforced by competitors.
Serious adverse events or other safety risks could require us to abandon development and preclude, delay or limit approval of our future product candidates, limit the scope of any approved label or market acceptance, or cause the recall or loss of marketing approval of products that are already marketed.
If any of our future product candidates prior to or after any approval for commercial sale, cause serious or unexpected side effects, or are associated with other safety risks such as misuse, abuse or diversion, a number of potentially significant negative consequences could result, including:
| ● | regulatory authorities may interrupt, delay or halt clinical trials; |
| ● | regulatory authorities may deny regulatory approval of our future product candidates; |
| ● | regulatory authorities may require certain labeling statements, such as warnings or contraindications or limitations on the indications for use, and/or impose restrictions on distribution in the form of a REMS in connection with approval or post-approval; |
| ● | regulatory authorities may withdraw their approval, require more onerous labeling statements, impose more restrictive REMS, or require us to recall any product that is approved; |
| ● | we may be required to change the way the product is administered or conduct additional clinical trials; |
| ● | our relationships with our collaboration partners may suffer; |
| ● | we could be sued and held liable for harm caused to patients; or |
| ● | our reputation may suffer. The reputational risk is heightened with respect to those of our future product candidates that are being developed for pediatric indications. |
We may voluntarily suspend or terminate our clinical trials if at any time we believe that the products present an unacceptable risk to participants, or if preliminary data demonstrates that our future product candidates are unlikely to receive regulatory approval or unlikely to be successfully commercialized. Following receipt of approval for commercial sale of a product, we may voluntarily withdraw or recall that product from the market if at any time we believe that its use, or a person’s exposure to it, may cause adverse health consequences or death. To date, we have not withdrawn, recalled or taken any other action, voluntary or mandatory, to remove an approved product from the market. In addition, regulatory agencies, IRBs or data safety monitoring boards may at any time recommend the temporary or permanent discontinuation of our clinical trials or request that we cease using investigators in the clinical trials if they believe that the clinical trials are not being conducted in accordance with applicable regulatory requirements, or that they present an unacceptable safety risk to participants. Although we have never been asked by a regulatory agency, IRB or data safety monitoring board to temporarily or permanently discontinue a clinical trial, if we elect or are forced to suspend or terminate a clinical trial of any of our future product candidates, the commercial prospects for that product will be harmed and our ability to generate product revenue from that product may be delayed or eliminated. Furthermore, any of these events may result in labeling statements such as warnings or contraindications. In addition, such events or labeling could prevent us or our partners from achieving or maintaining market acceptance of the affected product and could substantially increase the costs of commercializing our future product candidates and impair our ability to generate revenue from the commercialization of these products either by our company or by our collaboration partners.
72
The development of REMS for our future product candidates could cause delays in the approval process and would add additional layers of regulatory requirements that could impact our ability to commercialize our future product candidates in the U.S. and reduce their market potential.
Even if the FDA approves our NDA for any of our future product candidates without requiring a REMS as a condition of approval of the NDA, the FDA may, post-approval, require a REMS for any of our future product candidates if it becomes aware of new safety information that makes a REMS necessary to ensure that the benefits of the drug outweigh the potential risks. REMS elements can include medication guides, communication plans for health care professionals, and elements to assure safe use (“ETASU”). ETASU can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring and the use of patient registries. Moreover, product approval may require substantial post-approval testing and surveillance to monitor the drug’s safety or efficacy. We may be required to adopt a REMS for our future product candidates to ensure that the benefits outweigh the risks of abuse, misuse, diversion and other potential safety concerns. There can be no assurance that the FDA will approve a manageable REMS for our future product candidates, which could create material and significant limits on our ability to successfully commercialize our future product candidates in the U.S. Delays in the REMS approval process could result in delays in the NDA approval process. In addition, as part of the REMS, the FDA could require significant restrictions, such as restrictions on the prescription, distribution and patient use of the product, which could significantly impact our ability to effectively commercialize our future product candidates, and dramatically reduce their market potential, thereby adversely impacting our business, financial condition and results of operations. Even if initial REMS are not highly restrictive, if, after launch, our future product candidates were to be subject to significant abuse/non-medical use or diversion from illicit channels, this could lead to negative regulatory consequences, including a more restrictive REMS.
Risks Related to our Reliance Upon Third Parties
Our existing collaboration arrangements and any that we may enter into in the future may not be successful, which could adversely affect our ability to develop and commercialize our future product candidates.
We are a party to, and may seek additional, collaboration arrangements with pharmaceutical or biotechnology companies for the development or commercialization of our future product candidates. We may, with respect to our future product candidates, enter into new arrangements on a selective basis depending on the merits of retaining commercialization rights for ourselves as compared to entering into selective collaboration arrangements with leading pharmaceutical or biotechnology companies for each product candidate, both in the U.S. and internationally. To the extent that we decide to enter into collaboration agreements, we will face significant competition in seeking appropriate collaborators and the terms of any collaboration or other arrangements that we may establish may not be favorable to us.
Any existing or future collaboration that we enter into may not be successful. The success of our collaboration arrangements will depend heavily on the efforts and activities of our collaborators. Collaborators generally have significant discretion in determining the efforts and resources that they will apply to these collaborations. Disagreements between parties to a collaboration arrangement regarding development, intellectual property, regulatory or commercialization matters, can lead to delays in the development process or commercialization of the applicable product candidate and, in some cases, termination of the collaboration arrangement. These disagreements can be difficult to resolve if neither of the parties has final decision-making authority. Any such termination or expiration could harm our business reputation and may adversely affect it financially.
73
We expect to depend on a limited number of suppliers for materials and components in order to manufacture our future product candidates. The loss of these suppliers, or their failure to supply us on a timely basis, could cause delays in our current and future capacity and adversely affect our business.
We expect to depend on a limited number of suppliers for the materials and components required to manufacture our future product candidates. As a result, we may not be able to obtain sufficient quantities of critical materials and components in the future. A delay or interruption by our suppliers may also harm our business, results of operations and financial condition. In addition, the lead time needed to establish a relationship with a new supplier can be lengthy, and we may experience delays in meeting demand in the event we must switch to a new supplier. The time and effort to qualify for and, in some cases, obtain regulatory approval for a new supplier could result in additional costs, diversion of resources or reduced manufacturing yields, any of which would negatively impact our operating results. Our dependence on single-source suppliers exposes us to numerous risks, including the following: our suppliers may cease or reduce production or deliveries, they may be subject to government investigations and regulatory actions that limit or prevent production capabilities for an extended period of time, raise prices or renegotiate terms; our suppliers may become insolvent; we may be unable to locate a suitable replacement supplier on acceptable terms or on a timely basis, or at all; and delays caused by supply issues may harm our reputation, frustrate our customers and cause them to turn to our competitors for future needs.
Risks Related to our Intellectual Property
We may not be able to adequately protect our future product candidates or our proprietary technology in the marketplace.
Our success will depend, in part, on our ability to obtain patents, protect our trade secrets and operate without infringing on the proprietary rights of others. We rely upon a combination of patents, trade secret protection (i.e., know-how), and confidentiality agreements to protect the intellectual property of our future product candidates. The strengths of patents in the pharmaceutical field involve complex legal and scientific questions, and can be uncertain. Where appropriate, we seek patent protection for certain aspects of our products and technology. Filing, prosecuting and defending patents globally can be prohibitively expensive.
Our policy is to look to patent technologies with commercial potential in jurisdictions with significant commercial opportunities. However, patent protection may not be available for some of the products or technology we are developing. If we must spend significant time and money protecting, defending or enforcing our patents, designing around patents held by others or licensing, potentially for large fees, patents or other proprietary rights held by others, our business, results of operations and financial condition may be harmed. We may not develop additional proprietary products that are patentable. As of the date hereof, we have an extensive portfolio of patents, including many granted patents and patents pending approval.
The patent positions of pharmaceutical products are complex and uncertain. The scope and extent of patent protection for our future product candidates are particularly uncertain. Our future product candidates will be based on medicinal chemistry instead of cannabis plants. While we have sought patent protection, where appropriate, directed to, among other things, composition-of-matter for its specific formulations, their methods of use, and methods of manufacture, we do not have and will not be able to obtain composition of matter protection on these previously known CBD derivatives per se. We anticipate that the products we develop in the future will be based upon synthetic compounds we may discover. Although we have sought, and will continue to seek, patent protection in the U.S., Europe and other countries for our proprietary technologies, future product candidates, their methods of use, and methods of manufacture, any or all of them may not be subject to effective patent protection. If any of our products are approved and marketed for an indication for which we do not have an issued patent, our ability to use our patents to prevent a competitor from commercializing a non-branded version of our commercial products for that non-patented indication could be significantly impaired or even eliminated.
74
Publication of information related to our future product candidates by our company or others may prevent it from obtaining or enforcing patents relating to these products and product candidates. Furthermore, others may independently develop similar products, may duplicate our products, or may design around our patent rights. In addition, any of our issued patents may be opposed and/or declared invalid or unenforceable. If we fail to adequately protect our intellectual property, we may face competition from companies who attempt to create a generic product to compete with our future product candidates. We may also face competition from companies who develop a substantially similar product to our future product candidates that is not covered by any of our patents.
Many companies have encountered significant problems in protecting, defending and enforcing intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property rights, particularly those relating to pharmaceuticals, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
If third parties claim that intellectual property used by our company infringes upon their intellectual property, our operating profits could be adversely affected.
There is a substantial amount of litigation, both within and outside the U.S., involving patent and other intellectual property rights in the pharmaceutical industry. We may, from time to time, be notified of claims that we are infringing upon patents, trademarks, copyrights or other intellectual property rights owned by third parties, and we cannot provide assurances that other companies will not, in the future, pursue such infringement claims against us, our commercial partners or any third-party proprietary technologies we have licensed. If we were found to infringe upon a patent or other intellectual property right, or if we failed to obtain or renew a license under a patent or other intellectual property right from a third party, or if a third party from whom we were licensing technologies was found to infringe upon a patent or other intellectual property rights of another third party, we may be required to pay damages, including damages of up to three times the damages found or assessed, if the infringement is found to be willful, suspend the manufacture of certain products or reengineer or rebrand our products, if feasible, or we may be unable to enter certain new product markets. Any such claims could also be expensive and time consuming to defend and divert management’s attention and resources. Our competitive position could suffer as a result. In addition, if we have declined or failed to enter into a valid non-disclosure or assignment agreement for any reason, we may not own the invention or its intellectual property, and our products may not be adequately protected. Thus, we cannot guarantee that any of our future product candidates, or our commercialization thereof, does not and will not infringe any third party’s intellectual property.
If we are not able to adequately prevent disclosure of trade secrets and other proprietary information, the value of our technology and products could be significantly diminished.
We rely on trade secrets to protect our proprietary technologies, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. We rely in part on confidentiality agreements with current and former employees, consultants, outside scientific collaborators, sponsored researchers, contract manufacturers, vendors and other advisors to protect our trade secrets and other proprietary information. These agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. In addition, we cannot guarantee that we have executed these agreements with each party that may have or have had access to our trade secrets. Any party with whom we or they have executed such an agreement may breach that agreement and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches.
Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. Also, some courts inside and outside the United States are less willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent them, or those to whom they disclose such trade secrets, from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor or other third-party, our competitive position would be harmed.
75
Risks Related to Controlled Substances
Controlled substance legislation differs between countries, and legislation in certain countries may restrict or limit our ability to sell our future product candidates.
Most countries are parties to the Single Convention on Narcotic Drugs 1961 and the Convention on Psychotropic Substances 1971, which governs international trade and domestic control of narcotic substances, including cannabis extracts. Countries may interpret and implement their treaty obligations in a way that creates a legal obstacle to us obtaining marketing approval for our future products in those countries. These countries may not be willing or able to amend or otherwise modify their laws and regulations to permit our future products to be marketed, or achieving such amendments to the laws and regulations may take a prolonged period of time. In that case, we would be unable to market our future product candidates in those countries in the near future or perhaps at all.
The product candidates that we are developing may be subject to U.S. controlled substance laws and regulations and failure to comply with these laws and regulations, or the cost of compliance with these laws and regulations, may adversely affect the results of our business operations, both during clinical development and post approval, and our financial condition.
The product candidates that we are developing may contain controlled substances as defined in The United States Federal Controlled Substances Act of 1970 and the Controlled Substances Import and Export Act, as amended (“CSA”). Controlled substances that are pharmaceutical products are subject to a high degree of regulation under the CSA, which establishes, among other things, certain registration, manufacturing quotas, security, recordkeeping, reporting, import, export and other requirements administered by the DEA. The DEA classifies controlled substances into five schedules: Schedule I, II, III, IV or V substances. Schedule I substances by definition have a high potential for abuse, no currently “accepted medical use” in the U.S., lack accepted safety for use under medical supervision, and may not be prescribed, marketed or sold in the U.S. Pharmaceutical products approved for use in the U.S. which contain a controlled substance are listed as Schedule II, III, IV or V, with Schedule II substances considered to present the highest potential for abuse or dependence and Schedule V substances the lowest relative risk of abuse among such substances.
While cannabis is a Schedule I controlled substance, products approved for medical use in the U.S. that contain cannabis or cannabis extracts should be placed in Schedules II-V, since approval by the FDA satisfies the “accepted medical use” requirement. If and when any of our future product candidates receive FDA approval, the DEA will make a scheduling determination. If the FDA, the DEA or any foreign regulatory authority determines that our future product candidates may have potential for abuse, it may require us to generate more clinical or other data than we currently anticipate to establish whether or to what extent the substance has an abuse potential, which could increase the cost and/or delay the launch of that product.
Facilities conducting research, manufacturing, distributing, importing or exporting, or dispensing controlled substances must be registered (licensed) to perform these activities and have the security, control, recordkeeping, reporting and inventory mechanisms required by the DEA to prevent drug loss and diversion. All these facilities must renew their registrations annually, except dispensing facilities, which must renew every three years. The DEA conducts periodic inspections of certain registered establishments that handle controlled substances. Obtaining the necessary registrations may result in delay of the importation, manufacturing or distribution of our future products. Furthermore, failure to maintain compliance with the CSA, particularly non-compliance resulting in loss or diversion, can result in regulatory action that could have a material adverse effect on our business, financial condition and results of operations. The DEA may seek civil penalties, refuse to renew necessary registrations, or initiate proceedings to restrict, suspend or revoke those registrations. In certain circumstances, violations could lead to criminal proceedings.
76
Individual states have also established controlled substance laws and regulations. Although state-controlled substances laws often mirror federal law, because the states are separate jurisdictions, they may separately schedule our future product candidates as well. State scheduling may delay commercial sale of any product for which we obtain federal regulatory approval and adverse scheduling could have a material adverse effect on the commercial attractiveness of such product. We or our partners must also obtain separate state registrations, permits or licenses in order to be able to obtain, handle, and distribute controlled substances for clinical trials or commercial sale, and failure to meet applicable regulatory requirements could lead to enforcement and sanctions by the states in addition to those from the DEA or otherwise arising under federal law.
Because our products may be controlled substances in the U.S., to conduct clinical trials in the U.S., each of our research sites must submit a research protocol to the DEA and obtain and maintain a DEA researcher registration that will allow those sites to handle and dispense our products and to obtain product from our importer. If the DEA delays or denies the grant of a research registration to one or more research sites, the clinical trial could be significantly delayed, and we could lose clinical trial sites. The importer for the clinical trials must also obtain an importer registration and an import permit for each import.
The legislation on cannabis in the EU differs among the member states, as this area is not yet fully harmonized. In Germany, for example, cannabis is regulated as a controlled substance (Betäubungsmittel) and its handling requires specific authorization.
The legalization and use of medical and recreational cannabis in the U.S. and abroad may impact our business.
There is a substantial amount of change occurring in the U.S. regarding the use of medical and recreational cannabis products. While cannabis products not approved by the FDA are Schedule I substances as defined under federal law, and their possession and use is not permitted according to federal law (except for research purposes, under DEA registration), at least 36 states and the District of Columbia have enacted state laws to enable possession and use of cannabis for medical purposes, and at least 18 states and the District of Columbia for recreational purposes. The U.S. Farm Bill, which was passed in 2018, descheduled certain material derived from hemp plants with extremely low tetrahydrocannabinol (“THC”) content. Although our business is quite distinct from that of online and dispensary cannabis companies, future legislation authorizing the sale, distribution, use, and insurance reimbursement of non-FDA approved cannabis products could affect our business.
General Business Risks Relating to our Company
Changes in laws or regulations, or a failure to comply with any laws and regulations, may adversely affect our business, investments and results of operations.
We are subject to laws, regulations and rules enacted by national, regional and local governments. In particular, we are required to comply with certain SEC, NASDAQ and other legal or regulatory requirements. Compliance with, and monitoring of, applicable laws, regulations and rules may be difficult, time consuming and costly. Those laws, regulations and rules and their interpretation and application may also change from time to time and those changes could have a material adverse effect on our business, investments and results of operations. In addition, a failure to comply with applicable laws, regulations and rules, as interpreted and applied, could have a material adverse effect on our business and results of operations.
77
Certain of our executive officers and directors are now, and all of them may in the future become, affiliated with entities engaged in business activities similar to those conducted by us and, accordingly, may have conflicts of interest in determining to which entity a particular business opportunity should be presented.
Our executive officers and directors are, or may in the future become, affiliated with entities that are engaged in business activities similar to those that are conducted by us. Our officers and directors also may become aware of business opportunities which may be appropriate for presentation to us and the other entities to which they owe certain fiduciary or contractual duties. Accordingly, they may have conflicts of interest in determining whether a particular business opportunity should be presented to our company or to another entity. These conflicts may not be resolved in our favor and a potential opportunity may be presented to another entity prior to its presentation to us. Our Certificate of Incorporation provides that we renounce our interest in any corporate opportunity offered to any director or officer unless such opportunity is expressly offered to such person solely in his or her capacity as a director or officer of our company and such opportunity is one we are legally and contractually permitted to undertake and would otherwise be reasonable for us to pursue.
Our executive officers, directors, security holders and their respective affiliates may have competitive pecuniary interests that conflict with our interests.
We have not adopted a policy that expressly prohibits our directors, executive officers, security holders or affiliates from having a direct or indirect pecuniary or financial interest in any investment to be acquired or disposed of by us or in any transaction to which we are a party or have an interest. In fact, we may enter into a strategic transaction with a target business that is affiliated with our directors or executive officers. Nor do we have a policy that expressly prohibits any such persons from engaging for their own account in business activities of the types conducted by us. Accordingly, such persons or entities may have a conflict between their interests and ours. Certain of our officers and directors hold positions with companies which may be competitors of us. See also the biographies of our officers and directors incorporated by reference herein below under “Directors, Officers and Corporate Governance”.
The value of our existing intangible assets may become impaired, depending upon future operating results.
Our intangible assets were approximately $51.5 million as of December 31, 2021, representing approximately 81% of our total assets. We evaluate our intangible assets annually, or more often if there is a triggering event, to determine whether all or a portion of their carrying value may no longer be recoverable, in which case a charge to earnings may be necessary. Any future evaluations requiring an asset impairment charge for intangible assets would adversely affect future reported results of operations and stockholders’ equity, although such charges would not affect our cash flow.
Our outstanding warrants may have an adverse effect on the market price of our common stock.
In our IPO, we issued warrants to purchase 5,750,000 shares of common stock as part of the units offered in our IPO and, simultaneously with the closing of our IPO, we issued in a private placement an aggregate of 502,500 private placement warrants contained in the private placement units, each exercisable to purchase one-half of one share of common stock at $5.75 per half share. Additionally, (a) in February 2021, we sold warrants to purchase up to 2,564,000 shares of common stock with an exercise price of $5.00 per share, (b) in May 2021, we issued warrants to purchase up to 63,658 shares of common stock at an exercise price of $5.28 per share, (c) we issued certain warrants to purchase up to 25,000 shares of common stock at an exercise price of $7.07 per share in July 2021, and (d) in August 2021, we sold warrants to purchase up to 2,500,000 shares of common stock with an exercise price of $7.50 per share. Such warrants, if and when exercised, will increase the number of issued and outstanding shares of our common stock and potentially reduce the value of the outstanding shares of common stock.
78
Our business has been, and may continue to be, adversely affected by the COVID-19 pandemic.
In December 2019, a novel strain of coronavirus (COVID-19) was reported to have surfaced in Wuhan, China. In January 2020, COVID-19 spread to other parts of the word, including the United States and Europe, and efforts to contain its spread have intensified, with varying degrees of success. As a result, businesses have closed and limits have been placed on travel and everyday activities. The extent to which COVID-19 may impact our business will depend on future developments, which are highly uncertain and cannot be predicted with confidence, such as the duration of the outbreak, travel restrictions and social distancing in the United States and other countries, business closures or business disruptions, and the effectiveness of actions taken in the United States and other countries to contain and treat the disease. Should the COVID-19 pandemic continue, our plans could be delayed or interrupted. The spread of COVID-19 has also created global economic uncertainty, which may cause partners, suppliers and potential customers to closely monitor their costs and reduce their spending budget. The foregoing could materially adversely affect the clinical trials, supply chain, financial condition and financial performance of our company.
Enrollment of patients in our clinical trials, maintaining patients in our ongoing clinical trials, doing follow up visits with recruited patients and collecting data have been, and may continue to be, delayed or limited as certain of our clinical trial sites limit their onsite staff or temporarily close as a result of the COVID-19 pandemic and ongoing government restrictions. In addition, patients may not be able or willing to visit clinical trial sites for dosing or data collection purposes due to limitations on travel and physical distancing imposed or recommended by federal or state governments or patients’ reluctance to visit the clinical trial sites during the pandemic. These factors resulting from the COVID-19 pandemic could delay or prevent the anticipated readouts from our clinical trials, which could ultimately delay or prevent our ability to generate revenues and could have a material adverse effect on our results of operations.
Risks Related to our Common Stock and Warrants
The market price of our common stock has been extremely volatile and may continue to be volatile due to numerous circumstances beyond our control.
The market price of our common stock has fluctuated, and may continue to fluctuate, widely, due to many factors, some of which may be beyond our control. These factors include, without limitation:
| ● | “short squeezes”; |
| ● | comments by securities analysts or other third parties, including blogs, articles, message boards and social and other media; |
| ● | large stockholders exiting their position in our securities or an increase or decrease in the short interest in our securities; |
| ● | actual or anticipated fluctuations in our financial and operating results; |
| ● | risks and uncertainties associated with the ongoing COVID-19 pandemic; |
| ● | changes in foreign currency exchange rates; |
| ● | the commencement, enrollment or results of our planned or future clinical trials of our product candidates or those of our competitors; |
| ● | the success of competitive drugs or therapies; |
| ● | regulatory or legal developments in the United States and other countries; |
| ● | the success of competitive products or technologies; |
| ● | developments or disputes concerning patent applications, issued patents or other proprietary rights; |
| ● | the recruitment or departure of key personnel; |
| ● | the level of expenses related to our product candidates or clinical development programs; |
| ● | the results of our efforts to discover, develop, acquire or in-license additional product candidates; |
79
| ● | actual or anticipated changes in estimates as to financial results, development timelines or recommendations by securities analysts; |
| ● | our inability to obtain or delays in obtaining adequate drug supply for any approved drug or inability to do so at acceptable prices; |
| ● | disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technologies; |
| ● | significant lawsuits, including patent or stockholder litigation; |
| ● | variations in our financial results or those of companies that are perceived to be similar to us; |
| ● | changes in the structure of healthcare payment systems, including coverage and adequate reimbursement for any approved drug; |
| ● | market conditions in the pharmaceutical and biotechnology sectors; |
| ● | general economic, political, and market conditions and overall fluctuations in the financial markets in the United States and abroad; and |
| ● | investors’ general perception of us and our business. |
Stock markets in general and our stock price in particular have recently experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of those companies and our company. For example, on March 26, 2021, our common stock experienced an intra-day trading high of $10.60 per share and a low of $6.72 per share. In addition, during 2021, the sale prices of our common stock has ranged from a high of $13.05 per share (on April 13, 2021) to a low of $2.41 per share (on January 4, 2021). During this time, we have not experienced any material changes in our financial condition or results of operations that would explain such price volatility or trading volume. These broad market fluctuations may adversely affect the trading price of our securities. Additionally, these and other external factors have caused and may continue to cause the market price and demand for our common stock to fluctuate substantially, which may limit or prevent our stockholders from readily selling their shares of our common stock and may otherwise negatively affect the liquidity of our common stock.
Information available in public media that is published by third parties, including blogs, articles, message boards and social and other media may include statements not attributable to the Company and may not be reliable or accurate.
We are aware of a large volume of information being disseminated by third parties relating to our operations, including in blogs, message boards and social and other media. Such information as reported by third parties may not be accurate, may lead to significant volatility in our securities and may ultimately result in our common stock or other securities declining in value.
80
A significant number of our shares are eligible for sale and their sale or potential sale may depress the market price of our common stock.
Sales of a significant number of shares of our common stock in the public market could harm the market price of our common stock. Most of our common stock is available for resale in the public market, including (a) options to purchase 2,741,000 shares of common stock with a weighted average exercise price of $4.77 per share; and (b) warrants to purchase 11,153,908 shares of common stock with a weighted average exercise price of $9.06 per share. If a significant number of shares were sold, such sales would increase the supply of our common stock, thereby potentially causing a decrease in its price. Some or all of our shares of common stock may be offered from time to time in the open market pursuant to effective registration statements and/or compliance with Rule 144 (which was available starting November 6, 2021, subject to compliance with Rule 144, due to our status as a former “shell company”), which sales could have a depressive effect on the market for our shares of common stock. Subject to certain restrictions, a person who has held restricted shares for a period of six months may generally sell common stock into the market. The sale of a significant portion of such shares when such shares are eligible for public sale may cause the value of our common stock to decline in value.
There may not be sufficient liquidity in the market for our securities in order for investors to sell their shares. The market price of our common stock may continue to be volatile.
The market price of our common stock will likely continue to be highly volatile. Some of the factors that may materially affect the market price of our common stock are beyond our control, such as conditions or trends in the industry in which we operate or sales of our common stock. This situation is attributable to a number of factors, including the fact that we are a small company which is relatively unknown to stock analysts, stock brokers, institutional investors and others in the investment community that generate or influence sales volume, and that even if we came to the attention of such persons, they tend to be risk-averse and would be reluctant to follow an unproven company such as ours or purchase or recommend the purchase of our shares until such time as we became more seasoned and viable.
As a consequence, there may be periods of several days or more when trading activity in our shares is minimal or non-existent, as compared to a mature issuer which has a large and steady volume of trading activity that will generally support continuous sales without an adverse effect on share price. It is possible that a broader or more active public trading market for our common stock will not develop or be sustained, or that trading levels will not continue. These factors may materially adversely affect the market price of our common stock, regardless of our performance. In addition, the public stock markets have experienced extreme price and trading volume volatility. This volatility has significantly affected the market prices of securities of many companies for reasons frequently unrelated to the operating performance of the specific companies. These broad market fluctuations may adversely affect the market price of our common stock.
Risks Associated with Our Governing Documents and Delaware Law
Our Certificate of Incorporation provides for indemnification of officers and directors at our expense and limits their liability, which may result in a major cost to us and hurt the interests of our stockholders because corporate resources may be expended for the benefit of officers or directors.
Our Certificate of Incorporation provides for indemnification as follows: “To the fullest extent permitted by applicable law, the Corporation is authorized to provide indemnification of, and advancement of expenses to, such agents of the Corporation (and any other persons to which Delaware law permits the Corporation to provide indemnification) through Bylaw provisions, agreements with such agents or other persons, vote of stockholders or disinterested directors or otherwise, in excess of the indemnification and advancement otherwise permitted by Section 145 of the Delaware General Corporation Law, subject only to limits created by applicable Delaware law (statutory or non-statutory), with respect to actions for breach of duty to the Corporation, its stockholders and others.”
We have been advised that, in the opinion of the SEC, indemnification for liabilities arising under federal securities laws is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification for liabilities arising under federal securities laws, other than the payment by us of expenses incurred or paid by a director, officer or controlling person in the successful defense of any action, suit or proceeding, is asserted by a director, officer or controlling person in connection with our activities, we will (unless in the opinion of our counsel, the matter has been settled by controlling precedent) submit to a court of appropriate jurisdiction, the question whether indemnification by us is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue. The legal process relating to this matter if it were to occur is likely to be very costly and may result in us receiving negative publicity, either of which factors is likely to materially reduce the market and price for our shares.
81
Our Certificate of Incorporation contains a specific provision that limits the liability of our directors for monetary damages to the Company and the Company’s stockholders and requires us, under certain circumstances, to indemnify officers, directors and employees.
The limitation of monetary liability against our directors, officers and employees under Delaware law and the existence of indemnification rights to them may result in substantial expenditures by us and may discourage lawsuits against our directors, officers and employees.
Our Certificate of Incorporation contains a specific provision that limits the liability of our directors for monetary damages to the Company and the Company’s stockholders. We also have contractual indemnification obligations under our employment and engagement agreements with our executive officers and directors. The foregoing indemnification obligations could result in us incurring substantial expenditures to cover the cost of settlement or damage awards against our directors and officers, which the Company may be unable to recoup. These provisions and resultant costs may also discourage us from bringing a lawsuit against our directors and officers for breaches of their fiduciary duties and may similarly discourage the filing of derivative litigation by our stockholders against our directors and officers, even though such actions, if successful, might otherwise benefit us and our stockholders.
Our directors have the right to authorize the issuance of shares of preferred stock and additional shares of our common stock.
Our directors, within the limitations and restrictions contained in our Certificate of Incorporation and without further action by our stockholders, have the authority to issue shares of preferred stock from time to time in one or more series and to fix the number of shares and the relative rights, conversion rights, voting rights, and terms of redemption, liquidation preferences and any other preferences, special rights and qualifications of any such series. Any issuance of shares of preferred stock could adversely affect the rights of holders of our common stock. Should we issue additional shares of our common stock at a later time, each investor’s ownership interest in our stock would be proportionally reduced.
Anti-takeover provisions may impede the acquisition of the Company.
Certain provisions of the Delaware General Corporation Law (DGCL) have anti-takeover effects and may inhibit a non-negotiated merger or other business combination. These provisions are intended to encourage any person interested in acquiring control of us to negotiate with, and to obtain the approval of, our directors, in connection with such a transaction. As a result, certain of these provisions may discourage a future acquisition of the Company, including an acquisition in which the stockholders might otherwise receive a premium for their shares. In addition, we can also authorize “blank check” preferred stock, which could be issued by our board of directors without stockholder approval and may contain voting, liquidation, dividend and other rights superior to our common stock.
Compliance, Reporting and Listing Risks
We incur significant costs to ensure compliance with U.S. and NASDAQ Capital Market reporting and corporate governance requirements.
We incur significant costs associated with our public company reporting requirements and with applicable U.S. and NASDAQ Capital Market corporate governance requirements, including requirements under the Sarbanes-Oxley Act of 2002 and other rules implemented by the SEC and The NASDAQ Capital Market. The rules of The NASDAQ Capital Market include requiring us to maintain independent directors, comply with other corporate governance requirements and pay annual listing and stock issuance fees. All of such SEC and NASDAQ obligations require a commitment of additional resources including, but not limited, to additional expenses, and may result in the diversion of our senior management’s time and attention from our day-to-day operations. We expect all of these applicable rules and regulations to significantly increase our legal and financial compliance costs and to make some activities more time consuming and costly. We also expect that these applicable rules and regulations may make it more difficult and more expensive for us to obtain director and officer liability insurance and we may be required to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. As a result, it may be more difficult for us to attract and retain qualified individuals to serve on our board of directors or as executive officers.
82
We will continue to incur increased costs as a result of being a reporting company, and given our limited capital resources, such additional costs may have an adverse impact on our profitability.
We are an SEC-reporting company. The rules and regulations under the Exchange Act require reporting companies to provide periodic reports with interactive data files, which require that we engage legal, accounting and auditing professionals, and eXtensible Business Reporting Language (XBRL) and EDGAR (Electronic Data Gathering, Analysis, and Retrieval) service providers. The engagement of such services can be costly, and we may continue to incur additional losses, which may adversely affect our ability to continue as a going concern. In addition, the Sarbanes-Oxley Act of 2002, as well as a variety of related rules implemented by the SEC, have required changes in corporate governance practices and generally increased the disclosure requirements of public companies. For example, as a result of being a reporting company, we are required to file periodic and current reports and other information with the SEC and we have adopted policies regarding disclosure controls and procedures and regularly evaluate those controls and procedures.
The additional costs we continue to incur in connection with being a reporting company (expected to be several hundred thousand dollars per year) will continue to further stretch our limited capital resources. Due to our limited resources, we have to allocate resources away from other productive uses in order to continue to comply with our obligations as an SEC reporting company. Further, there is no guarantee that we will have sufficient resources to continue to meet our reporting and filing obligations with the SEC as they come due.
We may not be able to comply with NASDAQ’s continued listing standards.
Our common stock and warrants trade on The NASDAQ Capital Market under the symbols “ATNF” and “ATNFW,” respectively. Notwithstanding such listing, there can be no assurance any broker will be interested in trading our securities. Therefore, it may be difficult to sell your securities if you desire or need to sell them. There is also no guarantee that we will be able to maintain our listings on The NASDAQ Capital Market for any period of time by perpetually satisfying NASDAQ’s continued listing requirements. Our failure to continue to meet these requirements may result in our securities being delisted from NASDAQ.
Among the conditions required for continued listing on The NASDAQ Capital Market, NASDAQ requires us to maintain at least $2.5 million in stockholders’ equity, $35 million of market value of listed securities, or $500,000 in net income over the prior two years or two of the prior three years, to have a majority of independent directors, and to maintain a stock price over $1.00 per share. Our stockholders’ equity may not remain above NASDAQ’s $2.5 million minimum, our market value of listed securities may not remain above $35,000,000, we may not generate over $500,000 of yearly net income, we may not be able to maintain independent directors, and we may not be able to maintain a stock price over $1.00 per share. Furthermore, we are required to maintain a majority of independent directors and at least three members on our audit committee.
If we fail to comply with NASDAQ rules and requirements, our stock may be delisted. In addition, even if we demonstrate compliance with the requirements above, we will have to continue to meet other objective and subjective listing requirements to continue to be listed on The NASDAQ Capital Market. Delisting from The NASDAQ Capital Market could make trading our common stock and/or warrants more difficult for investors, potentially leading to declines in our share price and liquidity. Without a NASDAQ Capital Market listing, stockholders may have a difficult time getting a quote for the sale or purchase of our stock, the sale or purchase of our stock would likely be made more difficult and the trading volume and liquidity of our stock could decline. Delisting from The NASDAQ Capital Market could also result in negative publicity and could also make it more difficult for us to raise additional capital. The absence of such a listing may adversely affect the acceptance of our common stock and/or warrants as currency or the value accorded by other parties. Further, if we are delisted, we would also incur additional costs under state blue sky laws in connection with any sales of our securities. These requirements could severely limit the market liquidity of our common stock and/or warrants and the ability of our stockholders to sell our common stock and/or warrants in the secondary market. If our common stock and/or warrants are delisted by NASDAQ, our common stock and/or warrants may be eligible to trade on an over-the-counter quotation system, such as the OTCQB Market, where an investor may find it more difficult to sell our stock or obtain accurate quotations as to the market value of our common stock and/or warrants. In the event our common stock and/or warrants are delisted from The NASDAQ Capital Market, we may not be able to list our common stock and/or warrants on another national securities exchange or obtain quotation on an over-the counter quotation system.
83
We may experience adverse impacts on our reported results of operations as a result of adopting new accounting standards or interpretations.
Our implementation of and compliance with changes in accounting rules, including new accounting rules and interpretations, could adversely affect our reported financial position or operating results or cause unanticipated fluctuations in our reported operating results in future periods.
Risks Relating to The JOBS Act
The JOBS Act allows us to postpone the date by which we must comply with certain laws and regulations and to reduce the amount of information provided in reports filed with the SEC. We cannot be certain if the reduced disclosure requirements applicable to “emerging growth companies” will make our common stock less attractive to investors.
We are and we will remain an “emerging growth company” until the earliest to occur of (i) the last day of the fiscal year during which our total annual revenues equal or exceed $1.07 billion (subject to adjustment for inflation), (ii) the last day of the end of our 2022 fiscal year (5 years from our first public offering), (iii) the date on which we have, during the previous three-year period, issued more than $1 billion in non-convertible debt, or (iv) the date on which we are deemed a “large accelerated filer” (with at least $700 million in public float) under the Exchange Act. For so long as we remain an “emerging growth company” as defined in the JOBS Act, we may take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies” as described in further detail in the risk factors below. We cannot predict if investors will find our common stock less attractive because we will rely on some or all of these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile. If we avail ourselves of certain exemptions from various reporting requirements, as is currently our plan, our reduced disclosure may make it more difficult for investors and securities analysts to evaluate us and may result in less investor confidence.
Our election not to opt out of the JOBS Act extended accounting transition period may not make our financial statements easily comparable to other companies.
Pursuant to the JOBS Act, as an “emerging growth company”, we can elect to opt out of the extended transition period for any new or revised accounting standards that may be issued by the Public Company Accounting Oversight Board (PCAOB) or the SEC. We have elected not to opt out of such extended transition period which means that when a standard is issued or revised and it has different application dates for public or private companies, we, as an “emerging growth company”, can adopt the standard for the private company. This may make a comparison of our financial statements with any other public company which is not either an “emerging growth company” nor an “emerging growth company” which has opted out of using the extended transition period, more difficult or impossible as possible different or revised standards may be used.
84
The JOBS Act also allows us to postpone the date by which we must comply with certain laws and regulations intended to protect investors and to reduce the amount of information provided in reports filed with the SEC.
The JOBS Act is intended to reduce the regulatory burden on “emerging growth companies”. The Company meets the definition of an “emerging growth company” and so long as it qualifies as an “emerging growth company,” it will, among other things:
| ● | be exempt from the provisions of Section 404(b) of the Sarbanes-Oxley Act requiring that its independent registered public accounting firm provide an attestation report on the effectiveness of its internal control over financial reporting; | |
| ● | be exempt from the “say on pay” provisions (requiring a non-binding stockholder vote to approve compensation of certain executive officers) and the “say on golden parachute” provisions (requiring a non-binding stockholder vote to approve golden parachute arrangements for certain executive officers in connection with mergers and certain other business combinations) of The Dodd–Frank Wall Street Reform and Consumer Protection Act (Dodd-Frank Act) and certain disclosure requirements of the Dodd-Frank Act relating to compensation of Chief Executive Officers; | |
| ● | be permitted to omit the detailed compensation discussion and analysis from proxy statements and reports filed under the Exchange Act and instead provide a reduced level of disclosure concerning executive compensation; and | |
| ● | be exempt from any rules that may be adopted by the PCAOB requiring mandatory audit firm rotation or a supplement to the auditor’s report on the financial statements. |
The Company currently intends to take advantage of all of the reduced regulatory and reporting requirements that will be available to it so long as it qualifies as an “emerging growth company”. The Company has elected not to opt out of the extension of time to comply with new or revised financial accounting standards available under Section 102(b)(1) of the JOBS Act. Among other things, this means that the Company’s independent registered public accounting firm will not be required to provide an attestation report on the effectiveness of the Company’s internal control over financial reporting so long as it qualifies as an “emerging growth company”, which may increase the risk that weaknesses or deficiencies in the internal control over financial reporting go undetected. Likewise, so long as it qualifies as an “emerging growth company”, the Company may elect not to provide certain information, including certain financial information and certain information regarding compensation of executive officers, which it would otherwise have been required to provide in filings with the SEC, which may make it more difficult for investors and securities analysts to evaluate the Company. As a result, investor confidence in the Company and the market price of its common stock may be adversely affected.
Notwithstanding the above, we are also currently a “smaller reporting company”, meaning that we are not an investment company, an asset-backed issuer, or a majority-owned subsidiary of a parent company that is not a smaller reporting company and have a public float of less than $250 million and/or less than $100 million in annual revenues and a total public float less than $700 million during the most recently completed fiscal year. In the event that we are still considered a “smaller reporting company”, at such time as we cease being an “emerging growth company”, the disclosure we will be required to provide in our SEC filings will increase, but will still be less than it would be if we were not considered either an “emerging growth company” or a “smaller reporting company”. Specifically, similar to “emerging growth companies”, “smaller reporting companies” are able to provide simplified executive compensation disclosures in their filings; are exempt from the provisions of Section 404(b) of the Sarbanes-Oxley Act requiring that independent registered public accounting firms provide an attestation report on the effectiveness of internal control over financial reporting; and have certain other decreased disclosure obligations in their SEC filings, including, among other things, only being required to provide two years of audited financial statements in annual reports. Decreased disclosures in our SEC filings due to our status as an “emerging growth company” or “smaller reporting company” may make it harder for investors to analyze the Company’s results of operations and financial prospects.
85
General Risk Factors
Provisions in our Certificate of Incorporation and Delaware law may inhibit a takeover of us, which could limit the price investors might be willing to pay in the future for our common stock and could entrench management.
Our Certificate of Incorporation contains provisions that may discourage unsolicited takeover proposals that stockholders may consider to be in their best interests. These provisions include a staggered board of directors and the ability of the board of directors to designate the terms of and issue new series of preferred shares, which may make it more difficult for the removal of management and may discourage transactions that otherwise could involve payment of a premium over prevailing market prices for our securities. We are also subject to anti-takeover provisions under Delaware law, which could delay or prevent a change of control. Together, these provisions may make it more difficult for the removal of management and may discourage transactions that otherwise could involve payment of a premium over prevailing market prices for our securities.
Failure to adequately manage our planned aggressive growth strategy may harm our business or increase our risk of failure.
For the foreseeable future, we intend to pursue an aggressive growth strategy for the expansion of our operations through increased product development and marketing. Our ability to rapidly expand our operations will depend upon many factors, including our ability to work in a regulated environment, market value-added products effectively to independent pharmacies, establish and maintain strategic relationships with suppliers, and obtain adequate capital resources on acceptable terms. Any restrictions on our ability to expand may have a materially adverse effect on our business, results of operations, and financial condition. Accordingly, we may be unable to achieve our targets for sales growth, and our operations may not be successful or achieve anticipated operating results.
Additionally, our growth may place a significant strain on our managerial, administrative, operational, and financial resources and our infrastructure. Our future success will depend, in part, upon the ability of our senior management to manage growth effectively. This will require us to, among other things:
| ● | implement additional management information systems; | |
| ● | further develop our operating, administrative, legal, financial, and accounting systems and controls; |
| ● | hire additional personnel; | |
| ● | develop additional levels of management within our company; | |
| ● | locate additional office space; | |
| ● | maintain close coordination among our engineering, operations, legal, finance, sales and marketing, and client service and support organizations; and | |
| ● | manage our expanding international operations. |
As a result, we may lack the resources to deploy our services on a timely and cost-effective basis. Failure to accomplish any of these requirements could impair our ability to deliver services in a timely fashion or attract and retain new customers.
86
Our proprietary information, or that of our customers, suppliers and business partners, may be lost or we may suffer security breaches.
In the ordinary course of our business, we expect to collect and store sensitive data, including valuable and commercially sensitive intellectual property, clinical trial data, its proprietary business information and that of our future customers, suppliers and business partners, and personally identifiable information of our customers, clinical trial subjects and employees, patients, in its data centers and on our networks. The secure processing, maintenance and transmission of this information is critical to our operations. Despite our security measures, our information technology and infrastructure may be vulnerable to attacks by hackers or breached due to employee error, malfeasance or other disruptions. Any such breach could compromise our networks and the information stored there could be accessed, publicly disclosed, lost or stolen. Any such access, disclosure or other loss of information could result in legal claims or proceedings, liability under laws that protect the privacy of personal information, regulatory penalties, disrupt our operations, damage our reputation, and cause a loss of confidence in our products and our ability to conduct clinical trials, which could adversely affect our business and reputation and lead to delays in gaining regulatory approvals for our future product candidates. Although we maintain business interruption insurance coverage, our insurance might not cover all losses from any future breaches of our systems.
Failure of our information technology systems, including cybersecurity attacks or other data security incidents, could significantly disrupt the operation of our business.
Our business increasingly depends on the use of information technologies, which means that certain key areas such as research and development, production and sales are to a large extent dependent on our information systems or those of third-party providers. Our ability to execute our business plan and to comply with regulators’ requirements with respect to data control and data integrity, depends, in part, on the continued and uninterrupted performance of our information technology systems, or IT systems and the IT systems supplied by third-party service providers. As information systems and the use of software and related applications by our company, our business partners, suppliers, and customers become more cloud-based, there has been an increase in global cybersecurity vulnerabilities and threats, including more sophisticated and targeted cyber-related attacks that pose a risk to the security of our information systems and networks and the confidentiality, availability and integrity of data and information. In addition, our IT systems are vulnerable to damage from a variety of sources, including telecommunications or network failures, malicious human acts and natural disasters. Moreover, despite network security and backup measures, some of our servers are potentially vulnerable to physical or electronic break-ins, computer viruses and similar disruptive problems. Despite the precautionary measures we and our third-party service providers have taken to prevent unanticipated problems that could affect our IT systems, a successful cybersecurity attack or other data security incident could result in the misappropriation and/or loss of confidential or personal information, create system interruptions, or deploy malicious software that attacks our systems. It is also possible that a cybersecurity attack might not be noticed for some period of time. In addition, sustained or repeated system failures or problems arising during the upgrade of any of our IT systems that interrupt our ability to generate and maintain data, and in particular to operate our proprietary technology platform, could adversely affect our ability to operate our business. The occurrence of a cybersecurity attack or incident could result in business interruptions from the disruption of our IT systems, or negative publicity resulting in reputational damage with our stockholders and other stakeholders and/or increased costs to prevent, respond to or mitigate cybersecurity events. In addition, the unauthorized dissemination of sensitive personal information or proprietary or confidential information could expose us or other third-parties to regulatory fines or penalties, litigation and potential liability, or otherwise harm our business.
87
We may acquire other companies which could divert our management’s attention, result in additional dilution to our stockholders and otherwise disrupt our operations and harm our operating results.
We may in the future seek to acquire businesses, products or technologies that we believe could complement or expand our product offerings, enhance our technical capabilities or otherwise offer growth opportunities. The pursuit of potential acquisitions may divert the attention of management and cause us to incur various expenses in identifying, investigating and pursuing suitable acquisitions, whether or not they are consummated. If we acquire additional businesses, we may not be able to integrate the acquired personnel, operations and technologies successfully, effectively manage the combined business following the acquisition or realize anticipated cost savings or synergies. We also may not achieve the anticipated benefits from the acquired business due to a number of factors, including:
| ● | incurrence of acquisition-related costs; |
| ● | diversion of management’s attention from other business concerns; |
| ● | unanticipated costs or liabilities associated with the acquisition; |
| ● | harm to our existing business relationships with collaboration partners as a result of the acquisition; |
| ● | harm to our brand and reputation; |
| ● | the potential loss of key employees; |
| ● | use of resources that are needed in other parts of our business; and |
| ● | use of substantial portions of our available cash to consummate the acquisition. |
In the future, if our acquisitions do not yield expected returns, we may be required to take charges to our operating results arising from the impairment assessment process. Acquisitions may also result in dilutive issuances of equity securities or the incurrence of debt, which could adversely affect our operating results. In addition, if an acquired business fails to meet our expectations, our business, results of operations and financial condition may be adversely affected.
88
If we make any acquisitions, they may disrupt or have a negative impact on our business.
If we make acquisitions in the future, funding permitting, which may not be available on favorable terms, if at all, we could have difficulty integrating the acquired company’s assets, personnel and operations with our own. We do not anticipate that any acquisitions or mergers we may enter into in the future would result in a change of control of the Company. In addition, the key personnel of the acquired business may not be willing to work for us. We cannot predict the effect expansion may have on our core business. Regardless of whether we are successful in making an acquisition, the negotiations could disrupt our ongoing business, distract our management and employees and increase our expenses. In addition to the risks described above, acquisitions are accompanied by a number of inherent risks, including, without limitation, the following:
| ● | the difficulty of integrating acquired products, services or operations; | |
| ● | the potential disruption of the ongoing businesses and distraction of our management and the management of acquired companies; | |
| ● | difficulties in maintaining uniform standards, controls, procedures and policies; | |
| ● | the potential impairment of relationships with employees and customers as a result of any integration of new management personnel; | |
| ● | the potential inability or failure to achieve additional sales and enhance our customer base through cross-marketing of the products to new and existing customers; | |
| ● | the effect of any government regulations which relate to the business acquired; |
| ● | potential unknown liabilities associated with acquired businesses or product lines, or the need to spend significant amounts to retool, reposition or modify the marketing and sales of acquired products or operations, or the defense of any litigation, whether or not successful, resulting from actions of the acquired company prior to our acquisition; and | |
| ● | potential expenses under the labor, environmental and other laws of various jurisdictions. |
89
Our business could be severely impaired if and to the extent that we are unable to succeed in addressing any of these risks or other problems encountered in connection with an acquisition, many of which cannot be presently identified. These risks and problems could disrupt our ongoing business, distract our management and employees, increase our expenses and adversely affect our results of operations.
We may apply working capital and future funding to uses that ultimately do not improve our operating results or increase the value of our securities.
In general, we have complete discretion over the use of our working capital and any new investment capital we may obtain in the future. Because of the number and variety of factors that could determine our use of funds, our ultimate expenditure of funds (and their uses) may vary substantially from our current intended operating plan for such funds.
We intend to use existing working capital and future funding to support the development of our products and services, product purchases in our wholesale distribution division, the expansion of our marketing, or the support of operations to educate our customers. We will also use capital for market and network expansion, acquisitions, and general working capital purposes. However, we do not have more specific plans for the use and expenditure of our capital. Our management has broad discretion to use any or all of our available capital reserves. Our capital could be applied in ways that do not improve our operating results or otherwise increase the value of a stockholder’s investment.
We have never paid or declared any dividends on our common stock.
We have never paid or declared any dividends on our common stock or preferred stock. Likewise, we do not anticipate paying, in the near future, dividends or distributions on our common stock. Any future dividends on common stock will be declared at the discretion of our board of directors and will depend, among other things, on our earnings, our financial requirements for future operations and growth, and other facts as we may then deem appropriate. Since we do not anticipate paying cash dividends on our common stock, return on your investment, if any, will depend solely on an increase, if any, in the market value of our common stock.
Stockholders may be diluted significantly through our efforts to obtain financing and satisfy obligations through the issuance of additional shares of our common stock.
Wherever possible, our board of directors will attempt to use non-cash consideration to satisfy obligations. In many instances, we believe that the non-cash consideration will consist of restricted shares of our common stock or where shares are to be issued to our officers, directors and applicable consultants. Our board of directors has authority, without action or vote of the stockholders, but subject to NASDAQ rules and regulations (which generally require stockholder approval for any transactions which would result in the issuance of more than 20% of our then outstanding shares of common stock or voting rights representing over 20% of our then outstanding shares of stock), to issue all or part of the authorized but unissued shares of common stock. In addition, we may attempt to raise capital by selling shares of our common stock, possibly at a discount to market. These actions will result in dilution of the ownership interests of existing stockholders, which may further dilute common stock book value, and that dilution may be material. Such issuances may also serve to enhance existing management’s ability to maintain control of the Company because the shares may be issued to parties or entities committed to supporting existing management.
Our growth depends in part on the success of our strategic relationships with third parties.
In order to grow our business, we anticipate that we will need to continue to depend on our relationships with third parties, including our technology providers. Identifying partners, and negotiating and documenting relationships with them, requires significant time and resources. Our competitors may be effective in providing incentives to third parties to favor their products or services, or utilization of, our products and services. In addition, acquisitions of our partners by our competitors could result in a decrease in the number of our current and potential customers. If we are unsuccessful in establishing or maintaining our relationships with third parties, our ability to compete in the marketplace or to grow our revenue could be impaired and our results of operations may suffer. Even if we are successful, we cannot assure you that these relationships will result in increased customer use of our products or increased revenue.
90
Claims, litigation, government investigations, and other proceedings may adversely affect our business and results of operations.
We are currently subject to, and expect to continue to be regularly subject to, actual and threatened claims, litigation, reviews, investigations, and other proceedings. In addition, we have filed lawsuits against certain parties for matters we discovered which related to KBL, prior to the Business Combination. Any of these types of proceedings may have an adverse effect on us because of legal costs, disruption of our operations, diversion of management resources, negative publicity, and other factors. Our current legal proceedings are described in “Note 12 - Commitments and Contingencies”, under the heading “Litigation and Other Loss Contingencies”, in the consolidated financial statements included herein beginning on page F-1. The outcomes of these matters are inherently unpredictable and subject to significant uncertainties. Determining legal reserves and possible losses from such matters involves judgment and may not reflect the full range of uncertainties and unpredictable outcomes. Until the final resolution of such matters, we may be exposed to losses in excess of the amount recorded, and such amounts could be material. Should any of our estimates and assumptions change or prove to have been incorrect, it could have a material effect on our business, consolidated financial position, results of operations, or cash flows. In addition, it is possible that a resolution of one or more such proceedings, including as a result of a settlement, could require us to make substantial future payments, prevent us from offering certain products or services, require us to change our business practices in a manner materially adverse to our business, requiring development of non-infringing or otherwise altered products or technologies, damaging our reputation, or otherwise having a material effect on our operations.
For all of the foregoing reasons and others set forth herein, an investment in our securities involves a high degree of risk.
ITEM 1B. UNRESOLVED STAFF COMMENTS.
None.
None.
From time to time, we may be a party to litigation that arises in the ordinary course of our business.
Such current litigation or other legal proceedings are described in, and incorporated by reference in, this “Item 3. Legal Proceedings” of this Annual Report on Form 10-K from, “Note 12 - Commitments and Contingencies”, under the heading “Litigation and Other Loss Contingencies”, in the consolidated financial statements included herein beginning on page F-1. The Company believes that the resolution of currently pending matters will not individually or in the aggregate have a material adverse effect on our financial condition or results of operations. However, assessment of the current litigation or other legal claims could change in light of the discovery of facts not presently known to the Company or by judges, juries or other finders of fact, which are not in accord with management’s evaluation of the possible liability or outcome of such litigation or claims.
Additionally, the outcome of litigation is inherently uncertain. If one or more legal matters were resolved against the Company in a reporting period for amounts in excess of management’s expectations, the Company’s financial condition and operating results for that reporting period could be materially adversely affected.
ITEM 4. MINE SAFETY DISCLOSURES.
Not applicable.
91
ITEM 5. MARKET FOR THE REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our common stock, warrants, rights and units were previously listed on the NASDAQ Capital Market under the symbols “KBLM”, “KBLMW”, “KBLMR” and “KBLMU”, respectively. Our units commenced public trading on April 7, 2017 and our common stock, warrants and rights each commenced separate public trading on May 2, 2017. Our units automatically separated into the component securities upon consummation of the Business Combination and, as a result, no longer trade as a separate security, and our common stock and warrants began trading on the NASDAQ Capital Market under the symbols “ATNF” and “ATNFW,” respectively. Prior the Closing of the Business Combination, each unit consisted of one share of our common stock, one right convertible into 1/10th of one share of our common stock, and one warrant to purchase one half of one share of our common stock at an exercise price of $11.50 per whole share.
Holders
As of March 28, 2022, there were 34,087,244 shares of common stock issued and outstanding held by 69 holders of record, and 11,153,908 shares of common stock underlying 17,155,158 warrants outstanding to purchase shares of our common stock, with a weighted average exercise price of $9.06 per share, held by 17 holders of record.
Securities Authorized for Issuance Under Equity Compensation Plans
We have reserved 3,718,140 shares of our common stock for grant under our 2020 Omnibus Incentive Plan (“OIP”), of which 689,608 shares are available for future awards as of the date of this Report. The OIP is intended to be a vital component of our compensation program and the primary equity plan we use to grant equity-based incentive awards to our directors, officers, employees and consultants. Our Board believes that granting equity awards under the OIP will serve to align the interests of the key services providers of the Company and its subsidiaries with the Company’s stockholders, and that it would be in the best interest of the Company and its stockholders to make such grants.
Dividend Policy
We have never paid or declared any cash dividends on our common stock and do not anticipate paying cash dividends in the foreseeable future. We anticipate that we will retain all of our future earnings for use in the operation of our business and for general corporate purposes. Any determination to pay dividends in the future will be at the discretion of our board of directors. Accordingly, investors must rely on sales of their common stock after price appreciation, which may never occur, as the only way to realize any future gains on their investments.
Recent Sales of Unregistered Securities
None.
Issuer Purchases of Equity Securities
None.
Special Voting Shares
We have two classes of preferred stock designated, named our Class C Special Voting Shares and our Class K Special Voting Shares (collectively, the “Special Voting Shares”), with the rights and preferences specified below.
92
The Special Voting Shares have a par value of $0.0001 per share. The rights and preferences of each Special Voting Shares consists of the following:
| ● | the right to vote in all circumstances in which our common stock have the right to vote, with the common stock as one class; |
| ● | the Special Voting Shares entitle the holder Odyssey Trust Company (the Trustee) to an aggregate number of votes equal to the number of shares of common stock that were issuable to the holders of the previously outstanding shares of CannBioRex Purchaseco ULC and/or Katexco Purchaseco ULC, Canadian subsidiaries of 180 (the “Exchangeable Shares”); |
| ● | the holder of the Special Voting Shares (and, indirectly, the holders of the Exchangeable Shares) has the same rights as the holders of the common stock as to notices, reports, financial statements and attendance at all stockholder meetings; |
| ● | no entitlement to dividends; |
| ● | the holder of the Special Voting Shares is not entitled to any portion of any related distribution upon windup, dissolution or liquidation of the Company; and |
| ● | the Company may cancel the Special Voting Shares when there are no Exchangeable Shares outstanding and no option or other commitment of CannBioRex Purchaseco ULC and Katexco Purchaseco ULC which could require either CannBioRex Purchaseco ULC and Katexco Purchaseco ULC to issue more Exchangeable Shares. |
As set forth above, the holders of the Exchangeable Shares, through the applicable Special Voting Share, have voting rights and other attributes corresponding to the Common Stock. The Exchangeable Shares provide an opportunity for certain former Canadian resident holders of CBR Pharma or Katexco securities to obtain a deferral of taxable capital gains for Canadian income tax purposes in connection with the Reorganization.
As of the date of this Report, the Class C Special Voting Shares and our Class K Special Voting Shares have the right to vote 0 and 5,275 total voting shares, respectively.
A registrant such as the Company, that qualifies as a smaller reporting company, as defined by §229.10(f)(1), is not required to provide the information required by this Item.
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS.
The following discussion and analysis of the results of operations and financial condition of 180 Life Sciences Corp. as of and for the years ended December 31, 2021 and 2020 should be read in conjunction with our consolidated financial statements and the notes to those consolidated financial statements that are included elsewhere in this Annual Report. This Management’s Discussion and Analysis of Financial Condition and Results of Operations contains statements that are forward-looking. See “Cautionary Statement Regarding Forward-Looking Information” above. Actual results could differ materially because of the factors discussed in “Risk Factors” elsewhere in this Annual Report, and other factors that we may not know.
As of December 31, 2021, we had an accumulated deficit of $68,682,286 and net loss for the year ended December 31, 2021 of $20,324,648. As of December 31, 2021, we had a working capital deficit of $8,498,193. The accompanying consolidated financial statements have been prepared assuming the Company will continue as a going concern. As we are not generating revenues, we need to raise a significant amount of capital in order to pay our debts and cover our operating costs. While the Company raised money in August 2021, there is no assurance that we will be able to raise additional needed capital or that such capital will be available under favorable terms.
We are subject to all the substantial risks inherent in the development of a new business enterprise within an extremely competitive industry. Due to the absence of a long-standing operating history and the emerging nature of the markets in which we compete, we anticipate operating losses until we can successfully implement our business strategy, which includes all associated revenue streams. We may never ever achieve profitable operations or generate significant revenues.
We currently have a minimum monthly cash requirement spend of approximately $500,000. We believe that in the aggregate, we will require significant additional capital funding to support and expand the research and development and marketing of our products, fund future clinical trials, repay debt obligations, provide capital expenditures for additional equipment and development costs, payment obligations, office space and systems for managing the business, and cover other operating costs until our planned revenue streams from products are fully-implemented and begin to offset our operating costs, if ever.
Since our inception, we have funded our operations with the proceeds from equity and debt financings. We have experienced liquidity issues due to, among other reasons, our limited ability to raise adequate capital on acceptable terms. We have historically relied upon the issuance equity and promissory notes that are convertible into shares of our common stock to fund our operations and have devoted significant efforts to reduce that exposure. We anticipate that we will need to issue equity to fund our operations and repay our outstanding debt for the foreseeable future. If we are unable to achieve operational profitability or we are not successful in securing other forms of financing, we will have to evaluate alternative actions to reduce our operating expenses and conserve cash.
93
The accompanying consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. Accordingly, the consolidated financial statements do not include any adjustments relating to the recoverability of assets and classification of liabilities that might be necessary should the Company be unable to continue as a going concern. The consolidated financial statements included in this prospectus also include a going concern footnote.
Additionally, wherever possible, our Board of Directors will attempt to use non-cash consideration to satisfy obligations. In many instances, we believe that the non-cash consideration will consist of restricted shares of our common stock, preferred stock or warrants to purchase shares of our common stock. Our Board of Directors has authority, without action or vote of the shareholders, but subject to NASDAQ rules and regulations (which generally require shareholder approval for any transactions which would result in the issuance of more than 20% of our then outstanding shares of common stock or voting rights representing over 20% of our then outstanding shares of stock), to issue all or part of the authorized but unissued shares of common stock, preferred stock or warrants to purchase such shares of common stock. In addition, we may attempt to raise capital by selling shares of our common stock, possibly at a discount to market in the future. These actions will result in dilution of the ownership interests of existing shareholders, may further dilute common stock book value, and that dilution may be material. Such issuances may also serve to enhance existing management’s ability to maintain control of us, because the shares may be issued to parties or entities committed to supporting existing management.
Organization of MD&A
Our Management’s Discussion and Analysis of Financial Condition and Results of Operations (the “MD&A”) is provided in addition to the accompanying consolidated financial statements and notes to assist readers in understanding our results of operations, financial condition, and cash flows. MD&A is organized as follows:
| ● | Business Overview and Recent Events. A summary of the Company’s business and certain material recent events. |
| ● | Significant Financial Statement Components. A summary of the Company’s significant financial statement components. |
| ● | Results of Operations. An analysis of our financial results comparing the twelve months ended December 31, 2021 and 2020. |
| ● | Liquidity and Capital Resources. An analysis of changes in our balance sheets and cash flows and discussion of our financial condition. |
| ● | Critical Accounting Policies and Estimates. Accounting estimates that we believe are important to understanding the assumptions and judgments incorporated in our reported financial results and forecasts. |
Business Overview and Recent Events
On November 6, 2020 (“Closing Date”), the previously announced Business Combination was consummated following a special meeting of stockholders, where the stockholders of KBL considered and approved, among other matters, a proposal to adopt the Business Combination Agreement. Pursuant to the Business Combination Agreement, Merger Sub merged with 180, with 180 continuing as the surviving entity and becoming a wholly-owned subsidiary of KBL. As part of the Business Combination, KBL issued 17,500,000 shares of common stock and equivalents to the stockholders of 180, in exchange for all of the outstanding capital stock of 180. The Business Combination became effective November 6, 2020 and 180 filed a Certificate of Amendment of its Certificate of Incorporation in Delaware to change its name to 180 Life Corp., and KBL changed its name to 180 Life Sciences Corp.
This MD&A and the related financial statements for the year ended December 31, 2020 primarily covers the historical operations of 180 to the Closing Date (November 6, 2020) and then the combined operations of the two entities from the Closing Date to December 31, 2020. The Business Combination was accounted for as a reverse recapitalization with the assets and liabilities of KBL being consolidated commencing with the Closing Date. Thus, the results of operations for the year ended December 31, 2020, only include the combined results after the Closing Date. See Note 4 - Business Combination to the accompanying Consolidated Financial Statements included at the end of this Annual Report on Form 10-K.
Following the Closing of the Business Combination, we transitioned our operations to those of 180, which is a clinical stage biotechnology company headquartered in Palo Alto, California, focused on the development of therapeutics for unmet medical needs in chronic pain, inflammation, fibrosis and other inflammatory diseases, where anti-TNF therapy will provide a clear benefit to patients, by employing innovative research, and, where appropriate, combination therapy. We have three product development platforms:
| ● | fibrosis and anti-tumor necrosis factor (“TNF”); |
| ● | drugs which are derivatives of cannabidiol (“CBD”); and |
| ● | alpha 7 nicotinic acetylcholine receptor (“α7nAChR”). |
94
We have several future product candidates in development, including one product candidate which has recently completed a successful Phase 2b clinical trial in the United Kingdom for Dupuytren’s Contracture, a condition that affects the development of fibrous connective tissue in the palm of the hand. 180 was founded by several world-leading scientists in the biotechnology and pharmaceutical sectors.
We intend to invest resources to successfully complete the clinical programs that are underway, discover new drug candidates, and develop new molecules to build up on our existing pipeline to address unmet clinical needs. The product candidates are designed via a platform comprised of defined unit operations and technologies. This work is performed in a research and development environment that evaluates and assesses variability in each step of the process in order to define the most reliable production conditions.
We may rely on third-party contract manufacturing organizations (“CMOs”) and other third parties for the manufacturing and processing of the product candidates in the future. We believe the use of contract manufacturing and testing for the first clinical product candidates is cost-effective and has allowed us to rapidly prepare for clinical trials in accordance with our development plans. We expect that third-party manufacturers will be capable of providing and processing sufficient quantities of these product candidates to meet anticipated clinical trial demands.
COVID-19 Pandemic
In December 2019, a new strain of the coronavirus (COVID-19) was reported in Mainland China and during the first quarter of 2020 the virus had spread to over 150 countries, resulting in a global pandemic. This COVID-19 pandemic and the public health responses to contain it have resulted in global recessionary conditions, which did not exist at December 31, 2019. Among other effects, government-mandated closures, stay-at-home orders and other related measures have significantly impacted global economic activity and business investment in general. A continuation or worsening of the levels of market disruption and volatility seen in the recent past could have an adverse effect on our ability to access capital, and on our business, results of operations and financial condition. We have been closely monitoring the developments and have taken active measures to protect the health of our employees, their families, and our communities. The ultimate impact on the 2021 fiscal year and beyond will depend heavily on the duration of the COVID-19 pandemic and public health responses, including government-mandated closures, stay-at-home orders and social distancing mandates, as well as the substance and pace of macroeconomic recovery, all of which are uncertain and difficult to predict considering the rapidly evolving landscape of the COVID-19 pandemic and the public health responses to contain it.
The follow up time for patient data and the statistical analysis for the Phase 2b Dupuytren’s Contracture clinical trial was delayed as a result of COVID-19, but such follow-up and statistical analysis are now completed and the Company announced the top-line data results from the Phase 2b trial on December 1, 2021. Additionally, COVID-19 has delayed the initiation of certain clinical trials and may delay the initiation of other clinical trials in the future or otherwise have a material adverse effect on our future operations.
Close of Business Combination
On November 6, 2020 (the “Closing Date”), the Company consummated the previously announced Business Combination following a special meeting of stockholders held on November 5, 2020, where the stockholders of the Company considered and approved, among other matters, a proposal to adopt the Business Combination Agreement (as amended, the “Business Combination Agreement”), dated as of July 25, 2019. Pursuant to the Business Combination Agreement, among other things, a subsidiary of the Company merged with and into 180, with 180 continuing as the surviving entity and a wholly-owned subsidiary of the Company (the “Merger”). The Merger became effective on November 6, 2020. The Business Combination was accounted for as a reverse recapitalization of 180. All of 180’s capital stock outstanding immediately prior to the merger was exchanged for (i) 15,736,348 shares of 180LS common stock, (ii) 2 shares of Class C and Class K Special Voting Shares exchangeable into 1,763,652 shares of 180LS common stock which are presented as outstanding in the accompanying Statement of Changes in Stockholders’ Equity (Deficiency) due to the reverse recapitalization. The Company’s 6,928,645 outstanding shares of common stock are presented as being issued on the date of the Business Combination.
95
Significant Financial Statement Components
Research and Development
To date, 180’s research and development expenses have related primarily to discovery efforts and preclinical and clinical development of its three product platforms: fibrosis and anti-TNF; drugs which are derivatives of CBD, and α7nAChR. Research and development expenses consist primarily of costs associated with those three product platforms, which include:
| ● | expenses incurred under agreements with 180’s collaboration partners and third-party contract organizations, investigative clinical trial sites that conduct research and development activities on its behalf, and consultants; |
| ● | costs related to production of clinical materials, including fees paid to contract manufacturers; |
| ● | laboratory and vendor expenses related to the execution of preclinical and clinical trials; |
| ● | employee-related expenses, which include salaries, benefits and stock-based compensation; and |
| ● | facilities and other expenses, which include expenses for rent and maintenance of facilities, depreciation and amortization expense and other supplies. |
We expense all research and development costs in the periods in which they are incurred. We accrue for costs incurred as services are provided by monitoring the status of each project and the invoices received from our external service providers. We adjust our accrual as actual costs become known. When contingent milestone payments are owed to third parties under research and development arrangements or license agreements, the milestone payment obligations are expensed when the milestone results are achieved.
Research and development activities are central to our business model. Product candidates in later stages of clinical development generally have higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later-stage clinical trials. We expect that research and development expenses will increase over the next several years as clinical programs progress and as we seek to initiate clinical trials of additional product candidates. It is also expected that increased research and development expenses will be incurred as additional product candidates are selectively identified and developed. However, it is difficult to determine with certainty the duration and completion costs of current or future preclinical programs and clinical trials of product candidates.
96
The duration, costs and timing of clinical trials and development of product candidates will depend on a variety of factors that include, but are not limited to, the following:
| ● | per patient trial costs; |
| ● | the number of patients that participate in the trials; |
| ● | the number of sites included in the trials; |
| ● | the countries in which the trials are conducted; |
| ● | the length of time required to enroll eligible patients; |
| ● | the number of doses that patients receive; |
| ● | the drop-out or discontinuation rates of patients; |
| ● | potential additional safety monitoring or other studies requested by regulatory agencies; |
| ● | the impact of COVID-19 on the length of our trials; |
| ● | the duration of patient follow-up; and |
| ● | the efficacy and safety profile of the product candidates. |
In addition, the probability of success for each product candidate will depend on numerous factors, including competition, manufacturing capability and commercial viability. We will determine which programs to pursue and fund in response to the scientific and clinical success of each product candidate, as well as an assessment of each product candidate’s commercial potential.
Because the product candidates are still in clinical and preclinical development and the outcome of these efforts is uncertain, we cannot estimate the actual amounts necessary to successfully complete the development and commercialization of product candidates or whether, or when, we may achieve profitability. Due to the early-stage nature of these programs, we do not track costs on a project-by-project basis. As these programs become more advanced, we intend to track the external and internal cost of each program.
General and Administrative
General and administrative expenses consist primarily of salaries and other staff-related costs, including stock-based compensation for shares of common stock issued and options granted to founders, directors and personnel in executive, commercial, finance, accounting, legal, investor relations, facilities, business development and human resources functions and include vesting conditions.
97
Other significant general and administrative costs include costs relating to facilities and overhead costs, legal fees relating to corporate and patent matters, litigation, SEC filings, insurance, investor relations costs, fees for accounting and consulting services, and other general and administrative costs. General and administrative costs are expensed as incurred, and we accrue amounts for services provided by third parties related to the above expenses by monitoring the status of services provided and receiving estimates from our service providers and adjusting our accruals as actual costs become known.
It is expected that the general and administrative expenses will increase over the next several years to support our continued research and development activities, manufacturing activities, potential commercialization of our product candidates and the increased costs of operating as a public company. These increases are anticipated to include increased costs related to the hiring of additional personnel, developing commercial infrastructure, fees to outside consultants, lawyers and accountants, and increased costs associated with being a public company, as well as expenses related to services associated with maintaining compliance with Nasdaq listing rules and SEC requirements, insurance and investor relations costs.
Other Income
Other income primarily represents fees earned for research and development work performed for other companies, some of which are related parties.
Interest Expense
Interest expense consists primarily of interest expense related to debt instruments.
Gain (Loss) on Extinguishment of Convertible Notes
Gain (loss) on extinguishment of convertible notes represents the shortfall (excess) of the reacquisition cost of convertible notes as compared to their carrying value.
Change in Fair Value of Derivative Liabilities
Change in fair value of derivative liabilities represents the non-cash change in fair value of derivative liabilities during the reporting period. Gains resulting from change in fair value of derivative liabilities during the years ended December 31, 2021 and 2020, were driven by decreases in stock price during the period, resulting in a lower fair value of the underlying liability.
Offering Costs Allocated to Warrant Liabilities
Change in offering costs allocated to warrant liabilities represents placement agent fees and offering expenses which were allocated to the PIPE Warrants and expensed immediately as they are liability classified.
Change in Fair Value of Accrued Issuable Equity
Change in fair value of accrued issuable equity represents the non-cash change in fair value of accrued equity prior to its formal issuance.
98
CONSOLIDATED RESULTS OF OPERATIONS
Consolidated Results of Operations
For the Year Ended December 31, 2021 Compared to the Year Ended December 31, 2020
| For the Years Ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| Operating Expenses: | ||||||||
| Research and development | $ | 1,000,769 | $ | 2,217,371 | ||||
| Research and development - related parties | 2,947,536 | 75,633 | ||||||
| General and administrative | 11,230,118 | 3,169,260 | ||||||
| General and administrative - related parties | 462,580 | 185,848 | ||||||
| Total Operating Expenses | 15,641,003 | 5,648,112 | ||||||
| Loss From Operations | (15,641,003 | ) | (5,648,112 | ) | ||||
| Other (Expense) Income: | ||||||||
| Loss on sale and disposal of property and equipment | - | (37,174 | ) | |||||
| Gain on settlement of liabilities | 926,829 | - | ||||||
| Other income | (146,822 | ) | 15,334 | |||||
| Other income - related parties | - | 240,000 | ||||||
| Interest expense | (135,953 | ) | (1,002,424 | ) | ||||
| Interest expense - related parties | (50,255 | ) | (84,550 | ) | ||||
| Loss on extinguishment of convertible notes payable, net | (9,737 | ) | (2,580,655 | ) | ||||
| Change in fair value of derivative liabilities | (4,677,388 | ) | (1,816,309 | ) | ||||
| Change in fair value of accrued issuable equity | (9,405 | ) | 9,405 | |||||
| Offering costs allocated to warrant liabilities | (604,118 | ) | - | |||||
| Total Other Expense, Net | (4,706,849 | ) | (5,256,373 | ) | ||||
| Loss Before Income Taxes | (20,347,852 | ) | (10,904,485 | ) | ||||
| Income tax benefit | 23,204 | 20,427 | ||||||
| Net Loss | $ | (20,324,648 | ) | $ | (10,884,058 | ) | ||
Research and Development
During the year ended December 31, 2021, we incurred research and development expenses of $1,000,769 compared to $2,217,371 incurred for the year ended December 30, 2020, representing a decrease of $1,216,602 or 55%. The decrease includes a $90,000 decrease in research and development expenses related to drug discovery services provided by Evotec International GmbH in connection with a research and development agreement, a reduction in consulting expenses for the Scientific Advisory Board of $70,000, a decrease of $260,000 in research and development expenses related to the agreements amended and entered into with Yissum during 2020 and a decrease of $1,100,000 related to stock compensation paid to Yissum Research Development Company in 2020, offset by an increase in R&D tax credit of $500,000.
99
Research and Development – Related Parties
During the year ended December 31, 2021, we incurred research and development expenses – related parties of $2,947,536 compared to $75,633 incurred for the year ended December 31, 2020, representing an increase of 2,871,903 or 3,797%. The increase includes related party consultation fees paid to Jagdeep Nanchahal for his research in the Phase 2b clinical trial for Dupuytren’s Contracture (RIDD) totaling $800,000 and stock compensation expense paid to Mr. Nanchahal of approximately $1,400,000 and an additional overall increase in stock compensation expense of approximately $700,000.
General and Administrative
During the year ended December 31, 2021, we incurred general and administrative expenses of $11,230,118 compared to $3,169,260 incurred for the year ended December 31, 2020, representing an increase of $8,060,858 or 254%. The increase is attributable to an increase in professional fees of $2,800,000, which is comprised of legal fees of $1,600,000, penalties related to late filing of the Form S-1 for the February 2021 Offering of $525,000, fees of $275,000 related to the EarlyBird Settlement (see Note 12 – Commitments and Contingencies), and an increase in consulting fees of $700,000, which was offset by decreases in audit fees and merger expenses totaling $545,000. Salaries expense increased by approximately $830,000 due to increases in salaries expense, vacation expense, bonus expense and employee benefits expense resulting from hiring of additional personnel and accrued but unpaid bonuses. Stock-based compensation expense increased by $2,000,000 resulting from expense during 2021 for Directors and management. The prior year also had reversal of a bad debt provision of $1,700,000, while 2021 had no bad debt expense and there was in increase in D&O insurance of $1,100,000 for the year ended 2021.
General and Administrative – Related Parties
During the year ended December 31, 2021, we incurred general and administrative expenses – related parties of $462,580 compared to $185,848 incurred for the year ended December 31, 2020, representing an increase of $276,732, or 149%. The increase is primarily related to bad debt expense of $300,000 incurred in connection with a receivable from related parties.
Other (Expense) Income, Net
During the year ended December 31, 2021, we incurred other expenses, net of $4,706,849 compared to $5,256,373 for the year ended December 31, 2020, representing a decrease in other expenses of $549,524 or 10%. The decrease was primarily due to an increase of $2,861,079 in the change in fair value of derivative liabilities and an increase in warrant costs of $604,118 due to the offering, offset by a gain on the settlement of liabilities of $926,829 and a decrease in interest expense of $864,988 due to convertible debt being converted to stock. During the year ended December 31, 2021, there was a loss on the extinguishment of convertible notes payable of $9,737 compared to a loss of $2,580,655 during the year ended December 31, 2020.
Liquidity and Capital Resources
As of December 31, 2021 and 2020, we had cash balances of $8,224,508 and $2,108,544, respectively, and working capital deficits of $8,498,193 and $17,406,356, respectively.
For the years ended December 31, 2021 and 2020, cash used in operating activities was $19,371,428 and $3,871,961, respectively. Our cash used in operations for the year ended December 31, 2021 was primarily attributable to our net loss of $20,324,648, adjusted for non-cash expenses in the aggregate amount of $9,760,161, as well as $8,806,941 of net cash used in changes in the levels of operating assets and liabilities. A significant portion of cash used in operations during the year relates to $4.8 million of non-recurring expenses associated with the business combination (see Note 4). Our cash used in operations for the year ended December 31, 2020 was primarily attributable to our net loss of $10,884,058, adjusted for non-cash expenses in the aggregate amount of $4,679,931, as well as $2,332,166 of net cash provided by changes in the levels of operating assets and liabilities.
100
For the years ended December 31, 2021 and 2020, cash provided by investing activities was $0 and $14,490,724, respectively. Cash provided by investing activities during the year ended December 31, 2020 consisted of $10,280,739 of cash withdrawn from a trust account in connection with the Business Combination, $3,006,235 of cash acquired in the reverse recapitalization and $1,203,750 of proceeds received from the collection of notes receivable.
For the years ended December 31, 2021 and 2020, cash provided by (used in) financing activities was $25,411,919 and ($8,733,927), respectively. Cash provided by financing activities during the year ended December 31, 2021 was comprised of proceeds from the sale of common stock and warrants of $26,666,200 and proceeds from loans payable in the amount of $1,618,443, partially offset by repayments of convertible debt and loans payable of ($10,000) and ($807,594), respectively, and offering costs paid of ($2,055,130). The net cash used in financing activities during the year ended December 31, 2020 was due to payment of common stock redemptions in the amount of ($9,006,493), repayments made to related parties in the amount of ($201,859), and the repayment of loans in the amount of ($72,843), partially offset by sources of cash from the net proceeds from the sale of common stock in the amount of $72,500 and proceeds from loans and convertible notes in the amount of $474,768.
Our product candidates may never achieve commercialization and we anticipate that we will continue to incur losses for the foreseeable future. We expect that our research and development expenses, general and administrative expenses, and capital expenditures will continue to increase. As a result, until such time, if ever, as we are able to generate substantial product revenue, we expect to finance our cash needs through a combination of equity offerings, debt financings or other capital sources, including potentially collaborations, licenses and other similar arrangements, which may not be available on favorable terms, if at all. The sale of additional equity or debt securities, if accomplished, may result in dilution to our then stockholders. Our primary uses of capital are, and we expect will continue to be, compensation and related expenses, third-party clinical research and development services, license payments or milestone obligations that may arise, laboratory and related supplies, clinical costs, potential manufacturing costs, legal and other regulatory expenses and general overhead costs.
Our material cash requirements and time periods of such requirements from known contractual and other obligations include milestone and royalty payments related to license agreements with Oxford University and Yissum, payments related to the D&O insurance, payments to consultants and payments related to outside consulting firms, such as legal counsel, auditors, accountants, etc. These cash requirements, in the aggregate, amount to approximately $7,100,000 for 2022 and $33,400,000 for the years 2023 through 2026.
Further, our operating plans may change, and we may need additional funds to meet operational needs and capital requirements for clinical trials and other research and development activities. We currently have no credit facility or committed sources of capital. Because of the numerous risks and uncertainties associated with the development and commercialization of our product candidates, we are unable to estimate the amounts of increased capital outlays and operating expenditures associated with our current and anticipated product development programs.
101
We have not yet achieved profitability and expect to continue to incur cash outflows from operations. It is expected that our research and development and general and administrative expenses will continue to increase and, as a result, we will eventually need to raise additional capital to fund our operations. If we are unable to obtain adequate funds on reasonable terms, we may be required to significantly curtail or discontinue operations or obtain funds by entering into financing agreements on unattractive terms. Our operating needs include the planned costs to operate our business, including amounts required to fund working capital and capital expenditures. As of December 31, 2021, the conditions outlined above indicated that there was a substantial doubt about our ability to continue as a going concern within one year after the financial statement issuance date. However, in August 2021, the Company raised additional capital of approximately $13.88 million and with current cash on hand of $6,045,848 as of March 28, 2022, the Company expects to be able to continue as a going concern through the third quarter of 2022.
Our condensed consolidated financial statements have been prepared in conformity with accounting principles generally accepted in the United States of America (“U.S. GAAP”), which contemplate continuation of the Company as a going concern and the realization of assets and satisfaction of liabilities in the normal course of business. The carrying amounts of assets and liabilities presented in the condensed consolidated financial statements do not necessarily purport to represent realizable or settlement values. The condensed consolidated financial statements do not include any adjustment that might result from the outcome of this uncertainty.
Recent Financing and Settlement Transactions
February 2021 Offering
On February 19, 2021, the Company entered into a Securities Purchase Agreement with a number of institutional investors (the “Purchasers”) pursuant to which the Company agreed to sell to the Purchasers an aggregate of 2,564,000 shares (the “Shares”) of the Company’s common stock and warrants to purchase up to an aggregate of 2,564,000 shares of the Company’s common stock (the “SPA Warrants”), at a combined purchase price of $4.55 per Share and accompanying SPA Warrant (the “Offering”). Aggregate gross proceeds from the Offering were approximately $11.7 million, prior to deducting placement agent fees and estimated offering expenses payable by the Company. Net proceeds to the Company from the Offering, after deducting the placement agent fees and offering expenses payable by the Company, were approximately $10.8 million. The Offering closed on February 23, 2021.
Maxim Group LLC (the “Placement Agent”) acted as exclusive placement agent in connection with the Offering pursuant to an Engagement Letter between the Company and the Placement Agent dated January 26, 2021 (as amended on February 18, 2021). Pursuant to the Engagement Letter, the Placement Agent received a commission equal to seven percent (7%) of the aggregate gross proceeds of the Offering, or $816,634.
August 2021 Offering
On August 23, 2021, the Company entered into a Securities Purchase Agreement with certain purchasers (the “August 2021 Purchasers”), pursuant to which the Company agreed to sell an aggregate of 2,500,000 shares of common stock (the “August 2021 Shares”) and warrants to purchase up to an aggregate of 2,500,000 shares of common stock (the “August 2021 PIPE Warrants”), at a combined purchase price of $6.00 per share and August 2021 PIPE Warrant (the “August 2021 Offering”). Aggregate gross proceeds from the offering were approximately $15 million. Net proceeds to the Company from the offering, after deducting the placement agent fees and estimated offering expenses payable by the Company, were approximately $13.9 million. The placement agent fees and offering expenses were accounted for as a reduction of additional paid in capital. The Placement Agent received a commission equal to seven percent (7%) of the aggregate gross proceeds of the Offering, or $1,050,000. The offering closed on August 23, 2021.
102
In connection with the August 2021 Offering, the Company also entered into a Registration Rights Agreement, dated as of August 23, 2021, with the August 2021 Purchasers (the “August 2021 Registration Rights Agreement”). Pursuant to the August 2021 Registration Rights Agreement, the Company agreed to file a registration statement with the SEC on or prior to September 12, 2021 to register the resale of the August 2021 Shares and the shares of common stock issuable upon exercise of the August 2021 PIPE Warrants (the “Warrant Shares”), and to cause such registration statement to be declared effective on or prior to October 22, 2021 (or, in the event of a “full review” by the SEC, November 21, 2021). The registration statement was filed with the SEC on August 31, 2021 and the SEC declared it effective on September 9, 2021.
Conversion of Bridge Notes
On March 8, 2021, the holders of the Company’s convertible bridge notes, which were issued on December 27, 2019 and January 3, 2020 to various purchasers, converted an aggregate of $432,383, which included accrued interest of $66,633 owed under such convertible bridge notes, into an aggregate of 158,383 shares of common stock pursuant to the terms of such notes, as amended, at a conversion price of $2.73 per share.
Convertible Debt Conversions
From November 27, 2020 to February 5, 2021, the holders of the Company’s convertible promissory notes converted an aggregate of $4,782,107 owed under such convertible notes into an aggregate of 1,986,751 shares of common stock, pursuant to the terms of such notes, as amended, at conversion prices of between $2.00 and $3.29 per share.
During the third quarter of 2021, certain noteholders elected to convert certain convertible notes payable with an aggregate principal balance of $1,234,334 and an aggregate accrued interest balance of $105,850 into an aggregate of 467,123 shares of the Company’s common stock at conversion prices ranging from $2.45-$3.29 per share. The shares issued upon the conversion of the convertible promissory notes had a fair value at issuance of $1,941,125.
Earlybird Capital Settlement Agreement
On April 23, 2021, the Company settled the amounts due pursuant to a certain finder agreement entered into with EarlyBird Capital, Inc. (“EarlyBird”) on October 17, 2017 (the “Finder Agreement”). The Company’s Board of Directors determined it was in the best interests to settle all claims which had been made or could be made with respect to the Finder Agreement and entered into a settlement agreement (the “Settlement Agreement”). Pursuant to the Settlement Agreement, the Company paid EarlyBird a cash payment of $275,000 and issued 225,000 shares of the Company’s restricted common stock with a grant date value of $1,973,250 to EarlyBird, in full satisfaction of accounts payable in the amount of $1,750,000. The Company recorded a loss of $223,250 in connection with the Settlement Agreement, which is included in (loss) gain on settlement of liabilities in the accompanying condensed consolidated statements of operations.
Alpha Capital Settlement Agreement
On July 31, 2021, the Company reached an agreement to settle the amounts allegedly due pursuant to a certain convertible note agreement entered into with Alpha Capital Anstalt (“Alpha”) on September 8, 2020 (the “Alpha Note”). The Company’s Board of Directors determined it was in the best interest of the Company to settle all claims which had been made or could be made with respect to the Alpha Note and entered into a settlement agreement (“Alpha Settlement Agreement”). Pursuant to the Alpha Settlement Agreement, the Company issued 150,000 shares of common stock and three-year warrants to purchase 25,000 shares of the Company’s common stock at an exercise price of $7.07 per share, in exchange for full and complete satisfaction of the Alpha Note.
Exchanges of Related Party Loans and Convertible Notes
On September 30, 2021, Dr. Lawrence Steinman and Sir Marc Feldmann, Ph.D., each of whom serve as Co-Executive Chairmen of the Company’s Board of Directors, agreed with the Company to convert amounts owed under outstanding loans with an aggregate principal balance of $693,371 and an aggregate accrued interest balance of $157,741 into an aggregate of 141,852 shares of the Company’s common stock at the conversion price of $6.00 per share, pursuant to the terms of the agreement, which conversion rate was above the closing consolidated bid price of the Company’s common stock on the date the binding agreement was entered into. (See Note 10 - Loans Payable and Note 11 - Convertible Notes Payable for more information.)
103
Mintz Levin Settlement
In September 2021, the Company entered into a settlement agreement with Mintz, Levin, Cohn, Ferris, Glovsky and Popeo, P.C. (“Mintz”), whereby the Company agreed to pay $800,000 to Mintz for legal services rendered. Mintz had billed the Company an aggregate of $1,454,240 before factoring any interest charges. The Company recorded a gain of approximately $650,000 after making payment pursuant to the settlement agreement.
Cantor Fitzgerald & Co. Litigation Settlement
On October 12, 2021, the Company and Cantor Fitzgerald & Co. entered into a settlement agreement, whereby the Company agreed to pay to Cantor $200,000 in return for dismissal of the case against the Company. The Company sent the funds to Cantor on October 13, 2021. As of September 30, 2021, the Company recorded an accrual for the settlement amount as per the agreement.
On October 21, 2021, the Company received a notice of discontinuance and as a result, the matter between the Company and Cantor is settled and closed.
Critical Accounting Policies and Estimates
The Company’s consolidated financial statements are prepared in accordance with accounting principles that are generally accepted in the United States. The preparation of these consolidated financial statements requires management to make estimates and assumptions that affect the reported amounts of its assets, liabilities, revenue and expenses. The Company has identified certain policies and estimates as critical to its business operations and the understanding of its past or present results of operations related to (i) goodwill and (ii) intangible assets and in-process research and development (“IPR&D”). These policies and estimates are considered critical because they had a material impact, or they have the potential to have a material impact, on the Company’s consolidated financial statements and because they require management to make significant judgments, assumptions or estimates. The Company believes that the estimates, judgments and assumptions made when accounting for the items described below were reasonable, based on information available at the time they were made. However, actual results may differ from those estimates, and these differences may be material.
Goodwill/Intangible Assets and In-Process Research and Development (“IPR&D”)
The Company has a significant amount of goodwill, intangible assets and IPR&D assets that are assessed at least annually for impairment. At December 31, 2021, goodwill, intangible assets and IPR&D assets totaled $51.5 million, or 81%, of the Company’s total assets. The impairment analyses of these assets are considered critical because of their significance to the Company. Intangible assets arising from business combinations or acquisitions, such as goodwill, patents and IPR&D assets are initially recorded at estimated fair value. Licensed patents are amortized over the remaining life of the patent. IPR&D assets are considered to be indefinite-lived until the completion or abandonment of the associated research and development projects. Our goodwill was derived from acquisitions where the purchase price exceeded the fair value of the net assets acquired The Company is required to reassign goodwill to reporting units whenever reorganizations of the internal reporting structure change the composition of its reporting units. The Company identified one reporting unit which represents its sole operating segment.
The Company is required to assess goodwill/intangible assets and IPR&D assets at least annually, or more frequently, if an event occurs or circumstances change that indicates it is more likely than not the fair value of the Company’s reporting unit was less than its carrying value. In assessing goodwill/intangible assets and IPR&D assets for impairment, the Company may first assess qualitative factors to determine whether it is more likely than not that the fair value of its reporting unit is less than its carrying value. For December 31, 2021, the Company elected to bypass the qualitative analysis and proceeded directly to the two- step test.
The first step of the goodwill/intangible assets and IPR&D assets impairment test used to identify potential impairment compares the fair value of the reporting unit with its carrying amount, including goodwill/intangible assets and IPR&D assets. The Company determined the fair market value of its single reporting unit as of December 31, 2021 to be its market capitalization of $132,760,680, which represents $3.90 per share (the market close price) multiplied by 34,021,200 shares (consisting of 34,035,925 shares of common stock plus 5,275 special voting shares which are exchangeable into common stock for no additional consideration) on December 31, 2021. The carrying amount of the reporting unit as of December 31, 2021 was $40,207,861 (total assets of $63.5 million less total liabilities of $23.3 million).
Since the fair value of the Company ($132,760,680) exceeded the carrying value of the Company ($40,207,861) as of December 31, 2021, and the carrying value of the Company is greater than zero, management concluded the goodwill/intangible assets and IPR&D assets of the reporting unit was not impaired. The Company will continue to perform goodwill/intangible assets and IPR&D assets impairment testing on an annual basis, or as needed if there are changes to the composition of its reporting unit. For additional information on goodwill/intangible assets and IPR&D assets, see Note 3 – Summary of Significant Accounting Policies - Goodwill and Note 3 – Summary of Significant Accounting Policies - Intangible Assets and In-Process Research and Development.
104
Derivative Liabilities
The Company evaluates its debt and equity issuances to determine if those contracts or embedded components of those contracts qualify as derivatives requiring separate recognition in the Company’s financial statements. At December 31, 2021, derivative liabilities totaled $15.2 million, or 65%, of the Company’s total liabilities. The analyses of these liabilities are considered critical because of their significance to the Company. Entities must consider whether to classify contracts that may be settled in its own stock, such as warrants, as equity of the entity or as an asset or liability. If an event that is not within the entity’s control could require net cash settlement, then the contract should be classified as an asset or a liability rather than as equity.
The result of this accounting treatment is that the fair value of the embedded derivative is marked-to-market at each balance sheet date and recorded as a liability and the change in fair value is recorded in other (expense) income, net in the consolidated statements of operations. In circumstances where there are multiple embedded instruments that are required to be bifurcated, the bifurcated derivative instruments are accounted for as a single, compound derivative instrument. The classification of derivative instruments, including whether such instruments should be recorded as liabilities or as equity, is reassessed at the end of each reporting period. Equity instruments that are initially classified as equity that become subject to reclassification are reclassified to liability at the fair value of the instrument on the reclassification date. Derivative instrument liabilities are classified in the balance sheet as current or non-current based on whether or not net-cash settlement of the derivative instrument is expected within twelve months of the balance sheet date.
If the embedded conversion options do not require bifurcation, the Company then evaluates for the existence of a beneficial conversion feature by comparing the fair value of the Company’s underlying stock as of the commitment date to the effective conversion price of the instrument (the intrinsic value).
The Company has computed the fair value of warrants, options, convertible notes and convertible preferred stock issued using the Monte-Carlo and Black-Scholes option pricing models. The expected term used for warrants, convertible notes and convertible preferred stock are the contractual life and the expected term used for options issued is the estimated period of time that options granted are expected to be outstanding. The Company utilizes the “simplified” method to develop an estimate of the expected term of “plain vanilla” option grants. The Company is utilizing an expected volatility figure based on a review of the historical volatilities, over a period of time, equivalent to the expected life of the instrument being valued, of similarly positioned public companies within its industry. The risk-free interest rate was determined from the implied yields from U.S. Treasury zero-coupon bonds with a remaining term consistent with the expected term of the instrument being valued.
The Company evaluated the terms of its AGP warrants (see Note 9 – Derivative Liabilities) when they were originally earned and determined that the AGP Warrants should initially be liability-classified at their fair value at issuance with subsequent remeasurement (mark-to-market) at period ends. As of December 31, 2021, the Company has concluded that its warrants should remain liability-classified as of December 31, 2021due to the presence of the Tender Offer Provision combined with the existence of the Exchangeable Shares that have voting rights consistent with common stockholders. For additional information on derivative liabilities, see Note 3 – Summary of Significant Accounting Policies - Derivative Liabilities.
Recently Issued Accounting Pronouncements
See Note 3 – Summary of Significant Accounting Policies of our consolidated financial statements included within this Annual Report for a summary of recently issued and adopted accounting pronouncements.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK.
Pursuant to Item 305(e) of Regulation S-K (§ 229.305(e)), the Company is not required to provide the information required by this Item as it is a “smaller reporting company,” as defined by Rule 229.10(f)(1), however, the Company has provided the following information below relating to interest rate risk.
Interest Rate Risk
We are exposed to market risks in the ordinary course of its business. These risks primarily include interest rate sensitivities. As of December 31, 2021, we had $8,224,508 in cash and cash equivalents. We intend to hold our cash in interest-bearing money market accounts. Our primary exposure to market risk is interest rate sensitivity, which is affected by changes in the general level of U.S. interest rates. Due to the short-term maturities of our cash equivalents and the low risk profile of its investments, an immediate 100 basis point change in interest rates would not have a material effect on the fair market value of our cash equivalents.
105
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTAL DATA.
See “Index to Consolidated Financial Statements” which appears on page F-1 of this Annual Report on Form 10-K.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE.
None.
ITEM 9A. CONTROLS AND PROCEDURES.
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our principal executive officer and principal financial officer, evaluated the effectiveness of our disclosure controls and procedures, as such term is defined in Rules 13a-15(e) and 15d-15(e) promulgated under the Securities and Exchange Act of 1934, as amended (the "Exchange Act") as of December 31, 2021. Based on such evaluation, the principal executive officer and the principal financial officer have concluded that the Company's disclosure controls and procedures were effective.
Management’s Report on Internal Control over Financial Reporting
Management of 180 Life Sciences Corp. is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f). Internal control over financial reporting is a process designed by, or under the supervision of, our principal executive officer and principal financial officer, or persons performing similar functions, and effected by our board of directors to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that: (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the Company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the Company are being made only in accordance with authorizations of management and directors of the Company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the Company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions or that the degree of compliance with the policies or procedures may deteriorate.
We are a “smaller reporting company” as defined in Item 10(f)(1) of Regulation S-K under the Securities Act. For as long as we continue to be a smaller reporting company, we may take advantage of exemptions from various reporting requirements that are applicable to other public companies that are not smaller reporting companies. Additionally, this Report does not contain an attestation report of our registered public accounting firm regarding internal control over financial reporting since the Company, as an “emerging growth company,” is not required to provide such report.
Management of 180 Life Sciences Corp., including our principal financial officer, conducted an evaluation of the effectiveness of the Company’s internal control over financial reporting as of December 31, 2021 using the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”) in “Internal Control - Integrated Framework” (2013).
Management concluded that certain aspects of the Company’s internal control over financial reporting was not effective as of December 31, 2021, based on those criteria. Specifically, management’s conclusion was based on the following material weakness which existed as of December 31, 2020:
| ● | Financial Reporting Systems: The Company did not maintain a fully integrated financial consolidation and reporting system throughout the period and as a result, extensive manual analysis, reconciliation and adjustments were required in order to produce financial statements for external reporting purposes. |
The following material weakness which existed broadly as of December 31, 2020 has been narrowed on specific areas as of December 31, 2021 as the Company has made significant progress in remediating this material weakness:
| ● | Ineffective review controls over period end financial disclosure and reporting processes related to stock-based compensation and payroll expense classification. |
A material weakness is a control deficiency or combination of control deficiencies, that results in more than a remote likelihood that a material misstatement of the annual or interim financial statements will not be prevented or detected on a timely basis. As a company with limited accounting resources, a significant amount of management’s time and attention has been and will be diverted from our business to ensure compliance with these regulatory requirements.
106
Remediation Plan
Management intends to take steps to develop and enhance its internal controls over financial reporting, including:
| ● | Retaining the same accounting personnel throughout all reporting periods in 2022 to establish continuity of processes and implement sustainable improvements and efficiencies in the financial reporting and consolidation tools and procedures. |
| ● | Consider opportunities for improving the consolidations and financial statement processes, including exploring migrating to NetSuite, or a similar automated consolidations application to streamline the consolidations and reporting processes and enhance efficiency and accuracy. |
| ● | As part of the systems review and potential migration, our plan is to: |
| o | Strengthen the chart of accounts to provide required roll ups |
| o | Review current mapping and implement new procedures to enhance the controls on future changes |
| o | Automate reporting and calculations whenever possible |
| ● | To the extent manual processes, schedules and/or adjustments exist as part of, or following implementation, Management reviews must include additional high-level steps such as mapping considerations to financial reporting and detailed reviews of annual schedules to ensure the completeness and appropriate classification of expenses in the financial disclosure and reporting process. |
Remediation Of Material Weaknesses in Internal Control over Financial Reporting
The Company had previously reported that, as of December 31, 2020, it had identified the following two material weaknesses in its internal control over financial reporting:
| ● | Segregation of Duties: The Company does not currently have a sufficient complement of technical accounting and external reporting personnel commensurate to support standalone external financial reporting under public company or SEC requirements. Specifically, the Company did not effectively segregate certain accounting duties due to the small size of its accounting staff and maintain a sufficient number of adequately trained personnel necessary to anticipate and identify risks critical to financial reporting and the closing process. In addition, there were inadequate reviews and approvals by the Company’s personnel of certain reconciliations and other processes in day-to-day operations due to the lack of a full complement of accounting staff. |
| ● |
Insufficient written policies and procedures for accounting and financial reporting with respect to the requirements and application of GAAP and SEC disclosure requirements. |
During the year ended December 31, 2021, the Company has taken corrective action and/or placed in operation, controls to separately address the two material weaknesses described above, including Segregation of Duties, and Insufficient written policies and procedures for accounting and financial reporting. Over the course of the year, and concluding in the fourth quarter, management performed sufficient testing of the design and operating effectiveness of the applicable controls implemented to ensure sustainability. Upon the completion of testing, management concluded such controls have been operating effectively over a sufficient timeframe. These actions were subject to ongoing review by our Senior Management, as well as oversight by the Audit Committee of our Board of Directors.
Based on the corrective actions described above, and testing completed for the year ended December 31, 2021, it is management’s conclusion the two material weaknesses noted above that existed as of December 31, 2021 have been remediated.
The Company has also taken substantial corrective action and/or placed in operation, controls to address the material weakness related to the ineffective review controls and reporting processes, however such actions and controls were deemed insufficient to remediate this material weakness for the year ended December 31, 2021.
Changes in Internal Control over Financial Reporting
There have been no changes in the Company’s internal control over financial reporting through the date of this Report or during the quarter ended December 31, 2021, that materially affected, or is reasonably likely to materially affect, the Company’s internal control over financial reporting.
None.
ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS.
None.
107
Information required by Items 10, 11, 12, 13 and 14 of Part III is omitted from this Annual Report and will be filed in a definitive proxy statement or by an amendment to this Annual Report not later than 120 days after the end of the fiscal year covered by this Annual Report.
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE.
The information required by this Item is included under the headings “Election of Directors”, “Executive Officers”, “Corporate Governance”, “Code of Ethics”, “Board Committee Membership”, and “Delinquent Section 16(a) Reports” (to the extent applicable and warranted) in the Company’s 2022 Proxy Statement to be filed with the U.S. Securities and Exchange Commission (“SEC”) within 120 days after December 31, 2021, in connection with the solicitation of proxies for the Company’s 2022 annual meeting of stockholders and is incorporated herein by reference.
ITEM 11. EXECUTIVE COMPENSATION.
The information required by this Item is included under the headings “Executive and Director Compensation”, “Executive Compensation”, “Director Compensation”, “Outstanding Equity Awards at Fiscal Year-End”, “Compensation Committee Interlocks and Insider Participation” and “Compensation Committee Report” (to the extent required), in the Company’s 2022 Proxy Statement to be filed with the SEC within 120 days after December 31, 2021 and is incorporated herein by reference.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS.
The information required by this Item is included under the heading “Voting Rights and Principal Stockholders” and “Equity Compensation Plan Information” in the Company’s 2022 Proxy Statement to be filed with the SEC within 120 days after December 31, 2021, and is incorporated herein by reference.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE.
The information required by this Item is included under the heading “Voting Rights and Principal Stockholders” and “Equity Compensation Plan Information” in the Company’s 2022 Proxy Statement to be filed with the SEC within 120 days after December 31, 2021, and is incorporated herein by reference.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES.
Our independent public accounting firm is Marcum LLP, San Francisco, CA, PCAOB Auditor ID Auditor Firm Id: 688.
The information required by this Item is included under the heading “Ratification of Appointment of Auditors”-“Audit Fees” in the Company’s 2022 Proxy Statement to be filed with the SEC within 120 days after December 31, 2021 and is incorporated herein by reference.
108
ITEM 15. EXHIBITS, FINANCIAL STATEMENTS AND SCHEDULES
(a) Documents filed as part of this Annual Report:
The following is an index of the financial statements, schedules and exhibits included in this Form 10-K or incorporated herein by reference.
(1) All Financial Statements
(2) Consolidated Financial Statement Schedules
Except as provided above, all financial statement schedules have been omitted, since the required information is not applicable or is not present in amounts sufficient to require submission of the schedule, or because the information required is included in the consolidated financial statements and notes thereto included in this Form 10-K.
109
(3) Exhibits
EXHIBIT INDEX
110
111
112
113
114
| * |
Filed herewith.
|
| ** |
Furnished herewith.
|
| # |
Management contract or compensatory plans or arrangements.
|
| £ | Certain schedules and exhibits have been omitted pursuant to Item 601(a)(5) of Regulation S-K. A copy of any omitted schedule or exhibit will be furnished supplementally to the Securities and Exchange Commission upon request; provided, however that 180 Life Sciences Corp. may request confidential treatment pursuant to Rule 24b-2 of the Securities Exchange Act of 1934, as amended, for any schedule or exhibit so furnished. |
None.
115
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| 180 LIFE SCIENCES CORP. | ||
| Date: March 30, 2022 | /s/ James N. Woody | |
| By: | James N. Woody, Chief Executive Officer (Principal Executive Officer) | |
| Date: March 30, 2022 | /s/ Ozan Pamir | |
| By: | Ozan Pamir, Interim Chief Financial Officer (Principal Financial and Accounting Officer) | |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature | Title | Date | ||
| /s/ James N. Woody | Chief Executive Officer and Director | March 30, | ||
| James N. Woody | (Principal Executive Officer) | 2022 | ||
| /s/ Ozan Pamir | Interim Chief Financial Officer | March 30, | ||
| Ozan Pamir | (Principal Financial and Accounting Officer) | 2022 | ||
| /s/ Marc Feldmann | Co-Executive Chairman and Director | March 30, | ||
| Marc Feldmann | 2022 | |||
| /s/ Lawrence Steinman | Co-Executive Chairman and Director | March 30, | ||
| Lawrence Steinman | 2022 | |||
| /s/ Larry Gold | Director | March 30, | ||
| Larry Gold, Ph.D. | 2022 | |||
| /s/ Donald A. McGovern, Jr. | Lead Director | March 30, | ||
| Donald A. McGovern, Jr. | 2022 | |||
| /s/ Pamela G. Marrone | Director | March 30, | ||
| Pamela G. Marrone | 2022 | |||
| /s/ Francis Knuettel II | Director | March 30, | ||
| Francis Knuettel II | 2022 | |||
| /s/ Russell T. Ray | Director | March 30, | ||
| Russell T. Ray | 2022 | |||
| /s/ Teresa DeLuca | Director | March 30, | ||
| Teresa DeLuca | 2022 |
116
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
180 LIFE SCIENCES CORP. AND SUBSIDIARIES
CONSOLIDATED FINANCIAL STATEMENTS AS OF DECEMBER 31, 2021 AND 2020
TABLE OF CONTENTS
| Page | |
| REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM | F-2 |
CONSOLIDATED FINANCIAL STATEMENTS: |
|
Consolidated Balance Sheets as of December 31, 2021 and 2020 |
F-3 |
| F-4 | |
| F-6 | |
| Consolidated Statements of Cash Flows For the Years Ended December 31, 2021 and 2020 | F-7 |
| Notes to the Consolidated Financial Statements | F-9 |
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and Board of Directors of
180 Life Sciences Corp.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of 180 Life Sciences Corp. and Subsidiaries (the “Company”) as of December 31, 2021 and 2020, the related consolidated statements of operations and comprehensive loss, changes in stockholders’ equity and cash flows for each of the two years in the period ended December 31, 2021, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2021, and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2021, in conformity with accounting principles generally accepted in the United States of America.
Explanatory Paragraph – Going Concern
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As more fully described in Note 2 to the financial statements, the Company has a significant working capital deficiency, has incurred significant losses and needs to raise additional funds to meet its obligations and sustain its operations. These conditions raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 2. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (the “PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Marcum llp
Marcum llp
We have served as the Company’s auditor since 2019.
San Francisco, CA
March 30, 2022
F-2
180 LIFE SCIENCES CORP. AND SUBSIDIARIES
CONSOLIDATED BALANCE SHEETS
(Expressed in US Dollars)
| December 31, | December 31, | |||||||
| 2021 | 2020 | |||||||
| Assets | ||||||||
| Current Assets: | ||||||||
| Cash | $ | 8,224,508 | $ | 2,108,544 | ||||
| Due from related parties | - | 300,000 | ||||||
| Prepaid expenses and other current assets | 2,976,583 | 1,606,414 | ||||||
| Total Current Assets | 11,201,091 | 4,014,958 | ||||||
| Intangible assets, net | 1,948,913 | 2,047,818 | ||||||
| In-process research and development | 12,575,780 | 12,569,793 | ||||||
| Goodwill | 36,987,886 | 36,900,801 | ||||||
| Total Assets | $ | 62,713,670 | $ | 55,533,370 | ||||
| Liabilities and Stockholders’ Equity | ||||||||
| Current Liabilities: | ||||||||
| Accounts payable | $ | 586,611 | $ | 8,529,259 | ||||
| Accounts payable - related parties | - | 215,495 | ||||||
| Accrued expenses | 1,964,580 | 4,110,916 | ||||||
| Accrued expenses - related parties | 18,370 | 454,951 | ||||||
| Loans payable - current portion | 1,828,079 | 968,446 | ||||||
| Loans payable - related parties | 81,277 | 513,082 | ||||||
| Convertible notes payable | - | 1,916,195 | ||||||
| Convertible notes payable - related parties | - | 270,000 | ||||||
| Derivative liabilities | 15,220,367 | 4,442,970 | ||||||
| Total Current Liabilities | 19,699,284 | 21,421,314 | ||||||
| Accrued issuable equity | - | 43,095 | ||||||
| Loans payable - non current portion | 48,165 | 113,763 | ||||||
| Deferred tax liability | 3,643,526 | 3,668,329 | ||||||
| Total Liabilities | 23,390,975 | 25,246,501 | ||||||
| Commitments and contingencies (Note 12) | ||||||||
| Stockholders’ Equity: | ||||||||
| Preferred stock, $0.0001 par value; 5,000,000 shares authorized; (see designations and shares authorized for Series A, Class C and Class K preferred stock) | ||||||||
| Class C Preferred Stock; 1 share authorized, issued and outstanding at December 31, 2021 and 2020 | - | - | ||||||
| Class K Preferred Stock; 1 share authorized, issued and outstanding at December 31, 2021 and 2020 | - | - | ||||||
| Common stock, $0.0001 par value; 100,000,000 shares authorized; 34,035,925 and 26,171,225 shares issued and outstanding at December 31, 2021 and 2020, respectively | 3,404 | 2,617 | ||||||
| Additional paid-in capital | 107,184,137 | 78,005,004 | ||||||
| Accumulated other comprehensive income | 817,440 | 636,886 | ||||||
| Accumulated deficit | (68,682,286 | ) | (48,357,638 | ) | ||||
| Total Stockholders’ Equity | 39,322,695 | 30,286,869 | ||||||
| Total Liabilities and Stockholders’ Equity | $ | 62,713,670 | $ | 55,533,370 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
F-3
180 LIFE SCIENCES CORP. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
(Expressed in US Dollars)
| For the Year Ended | ||||||||
| December 31, | ||||||||
| 2021 | 2020 | |||||||
| Operating Expenses: | ||||||||
| Research and development | $ | 1,000,769 | $ | 2,217,371 | ||||
| Research and development - related parties | 2,947,536 | 75,633 | ||||||
| General and administrative | 11,230,118 | 3,169,260 | ||||||
| General and administrative - related parties | 462,580 | 185,848 | ||||||
| Total Operating Expenses | 15,641,003 | 5,648,112 | ||||||
| Loss From Operations | (15,641,003 | ) | (5,648,112 | ) | ||||
| Other (Expense) Income: | ||||||||
| Loss on sale and disposal of property and equipment | - | (37,174 | ) | |||||
| Gain on settlement of liabilities | 926,829 | - | ||||||
| Other income | (146,822 | ) | 15,334 | |||||
| Other income - related parties | - | 240,000 | ||||||
| Interest expense | (135,953 | ) | (1,002,424 | ) | ||||
| Interest expense - related parties | (50,255 | ) | (84,550 | ) | ||||
| Loss on extinguishment of convertible notes payable, net | (9,737 | ) | (2,580,655 | ) | ||||
| Change in fair value of derivative liabilities | (4,677,388 | ) | (1,816,309 | ) | ||||
| Change in fair value of accrued issuable equity | (9,405 | ) | 9,405 | |||||
| Offering costs allocated to warrant liabilities | (604,118 | ) | - | |||||
| Total Other Expense, Net | (4,706,849 | ) | (5,256,373 | ) | ||||
| Loss Before Income Taxes | (20,347,852 | ) | (10,904,485 | ) | ||||
| Income tax benefit | 23,204 | 20,427 | ||||||
| Net Loss | (20,324,648 | ) | (10,884,058 | ) | ||||
| Deemed dividend related to the Series A Convertible Preferred Stock | - | (1,122,702 | ) | |||||
| Net Loss Attributable to Common Stockholders | $ | (20,324,648 | ) | $ | (12,006,760 | ) | ||
| Net Loss | $ | (20,324,648 | ) | $ | (10,884,058 | ) | ||
| Other Comprehensive Income: | ||||||||
| Foreign currency translation adjustments | 180,554 | 484,083 | ||||||
| Total Comprehensive Income | $ | (20,144,094 | ) | $ | (10,399,975 | ) | ||
| Basic and Diluted Net Loss per Common Share | $ | (0.65 | ) | $ | (0.66 | ) | ||
| Weighted Average Number of Common Shares Outstanding: | 31,355,439 | 18,154,056 | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-4
180 LIFE SCIENCES CORP. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY
(Expressed in US Dollars)
| For The Year Ended December 31, 2021 | ||||||||||||||||||||||||
| Common Stock | Additional Paid-in | Accumulated Other Comprehensive | Accumulated | Total Stockholders’ | ||||||||||||||||||||
| Shares | Amount | Capital | Income | Deficit | Equity | |||||||||||||||||||
| Balance - January 1, 2021 | 26,171,225 | $ | 2,617 | $ | 78,005,004 | $ | 636,886 | $ | (48,357,638 | ) | $ | 30,286,869 | ||||||||||||
| Shares issued upon conversion of KBL debt (Note 11) | 467,123 | 47 | 1,941,078 | - | - | 1,941,125 | ||||||||||||||||||
| Shares issued upon conversion of 180 debt (Note 11) | 158,383 | 16 | 432,367 | - | - | 432,383 | ||||||||||||||||||
| Shares issued in connection with the financing, net of financing costs (Note 9) | 2,564,000 | 256 | 10,730,814 | - | - | 10,731,070 | ||||||||||||||||||
| Offering costs allocated to warrant liabilities (Note 13) | - | - | 604,118 | - | - | 604,118 | ||||||||||||||||||
| Warrants issued in connection with private offering, reclassified to derivative liabilities (Note 9) | - | - | (7,294,836 | ) | - | - | (7,294,836 | ) | ||||||||||||||||
| Shares issued upon exchange of common stock equivalents (Note 13) | 1,745,054 | 174 | (174 | ) | - | - | - | |||||||||||||||||
| Shares issued to settle accounts payable (Note 12) | 225,000 | 23 | 1,973,227 | - | - | 1,973,250 | ||||||||||||||||||
| Shares issued in connection with the August 2021 Offering, net of financing costs (Note 13) | 2,500,000 | 250 | 13,879,750 | - | - | 13,880,000 | ||||||||||||||||||
| Shares issued to settle convertible debt and derivative liabilities with Alpha Capital (Note 11) | 150,000 | 15 | 1,060,485 | - | - | 1,060,500 | ||||||||||||||||||
| Shares issued in connection with the repayment of related party loans and convertible notes (Note 13) | 141,852 | 15 | 851,097 | - | - | 851,112 | ||||||||||||||||||
| Stock based compensation (Note 13): | ||||||||||||||||||||||||
| Common stock | 317,553 | 31 | 2,148,858 | - | - | 2,148,889 | ||||||||||||||||||
| Options | - | - | 2,852,309 | - | - | 2,852,309 | ||||||||||||||||||
| Shares Cancelled | (404,265 | ) | (40 | ) | 40 | - | - | - | ||||||||||||||||
| Comprehensive income (loss): | ||||||||||||||||||||||||
| Net loss | - | - | - | - | (20,324,648 | ) | (20,324,648 | ) | ||||||||||||||||
| Other comprehensive income | - | - | - | 180,554 | - | 180,554 | ||||||||||||||||||
| Balance - December 31, 2021 | 34,035,925 | $ | 3,404 | $ | 107,184,137 | $ | 817,440 | $ | (68,682,286 | ) | $ | 39,322,695 | ||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-5
180 LIFE SCIENCES CORP. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY, continued
(Expressed in US Dollars)
| For The Year Ended December 31, 2020 | ||||||||||||||||||||||||
| Common Stock | Additional Paid-in | Accumulated Other Comprehensive | Accumulated | Total Stockholders’ | ||||||||||||||||||||
| Shares | Amount | Capital | Income | Deficit | Equity | |||||||||||||||||||
| Balance - January 1, 2020 | 13,846,925 | $ | 1,384 | $ | 75,890,295 | $ | 152,803 | $ | (37,473,580 | ) | $ | 38,570,902 | ||||||||||||
| Common stock issued for cash (Note 13) | 12,292 | 1 | 72,499 | - | - | 72,500 | ||||||||||||||||||
| Shares issued upon conversion of KBL debt (Note 11) | 1,519,628 | 152 | 4,164,833 | - | - | 4,164,985 | ||||||||||||||||||
| Shares issued upon conversion of 180 debt (Note 11) | 482,894 | 48 | 2,117,270 | 2,117,318 | ||||||||||||||||||||
| Shares issued upon the conversion of the Series A convertible preferred stock (Note 13) | 1,619,144 | 162 | 4,348,873 | 4,349,035 | ||||||||||||||||||||
| Shares issued upon exchange of common stock equivalents (Note 13) | 1,521,157 | 153 | (153 | ) | - | - | - | |||||||||||||||||
| Beneficial conversion feature on convertible debt issued (Note 11) | - | - | 329,300 | - | - | 329,300 | ||||||||||||||||||
| Deemed dividend on Series A convertible preferred stock (Note 13): | ||||||||||||||||||||||||
| Extinguishment loss | - | - | (565,659 | ) | - | - | (565,659 | ) | ||||||||||||||||
| Make-whole dividend | - | - | (333,333 | ) | - | - | (333,333 | ) | ||||||||||||||||
| Beneficial conversion feature | - | - | (223,710 | ) | - | - | (223,710 | ) | ||||||||||||||||
| Stock based compensation (Note 13): | ||||||||||||||||||||||||
| Common stock | 240,540 | 24 | 1,057,965 | - | - | 1,057,989 | ||||||||||||||||||
| Options | - | - | 7,798 | - | - | 7,798 | ||||||||||||||||||
| Effect of reverse recapitalization, net of cash acquired (Note 4) | 6,928,645 | 693 | (8,860,974 | ) | - | - | (8,860,281 | ) | ||||||||||||||||
| Comprehensive loss: | ||||||||||||||||||||||||
| Net loss | - | - | - | - | (10,884,058 | ) | (10,884,058 | ) | ||||||||||||||||
| Other comprehensive loss | - | - | - | 484,083 | - | 484,083 | ||||||||||||||||||
| Balance - December 31, 2020 | 26,171,225 | $ | 2,617 | $ | 78,005,004 | $ | 636,886 | $ | (48,357,638 | ) | $ | 30,286,869 | ||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-6
180 LIFE SCIENCES CORP. AND SUBSIDIARIES
CONDENSED CONSOLIDATED STATEMENTS OF CASH FLOWS
(Expressed in US Dollars)
| For the Years Ended | ||||||||
| December 31, | ||||||||
| 2021 | 2020 | |||||||
| Cash Flows From Operating Activities | ||||||||
| Net loss | $ | (20,324,648 | ) | $ | (10,884,058 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Stock-based compensation | ||||||||
| Shares issued for services | 2,148,889 | - | ||||||
| Amortization of stock options | 2,852,309 | 1,118,286 | ||||||
| Amortization of debt discount | - | 356,179 | ||||||
| Depreciation and amortization | 109,947 | 125,333 | ||||||
| Bad debt expense (recovery) - related parties | 300,000 | (1,699,825 | ) | |||||
| Gain on exchange rate transactions | - | 4,760 | ||||||
| Interest capitalized to debt principal | - | 396,535 | ||||||
| Gain on settlement of liabilities, net | (926,829 | ) | (25,643 | ) | ||||
| Loss on extinguishment of convertible note payable | 9,737 | 2,580,655 | ||||||
| Deferred tax benefit | (24,803 | ) | (20,427 | ) | ||||
| Offering costs allocated to warrant liabilities | 604,118 | - | ||||||
| Loss on sale/disposal of property and equipment | - | 37,174 | ||||||
| Change in fair value of derivative liabilities | 4,677,388 | 1,816,309 | ||||||
| Change in fair value of accrued issuable equity | 9,405 | (9,405 | ) | |||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid expenses and other current assets | (1,377,247 | ) | (210,195 | ) | ||||
| Due from related parties | - | (217,939 | ) | |||||
| Accounts payable | (5,730,537 | ) | 2,226,814 | |||||
| Accrued expenses | (1,646,657 | ) | 533,486 | |||||
| Accrued issuable equity | (52,500 | ) | - | |||||
| Total adjustments | 953,220 | 7,012,097 | ||||||
| Net Cash Used In Operating Activities | (19,371,428 | ) | (3,871,961 | ) | ||||
| Cash Flows From Investing Activities | ||||||||
| Cash withdrawn from Trust Account | - | 10,280,739 | ||||||
| Cash acquired in reverse merger | - | 3,006,235 | ||||||
| Proceeds from repayment of notes receivable | - | 1,203,750 | ||||||
| Net Cash Provided by Investing Activities | - | 14,490,724 | ||||||
| Cash Flows From Financing Activities | ||||||||
| Proceeds from sale of common stock and warrants | 26,666,200 | 72,500 | ||||||
| Offering costs in connection with sale of common stock and warrants | (2,055,130 | ) | - | |||||
| Repayment of advances from related party | - | (201,859 | ) | |||||
| Repayment of convertible debt | (10,000 | ) | - | |||||
| Repayment of loans payable | (807,594 | ) | (72,843 | ) | ||||
| Payment of common stock redemptions payable | - | (9,006,493 | ) | |||||
| Proceeds from loans payable | 1,618,443 | 275,049 | ||||||
| Proceeds from convertible notes payable | - | 82,500 | ||||||
| Proceeds from Paycheck Protection Program Loan | - | 53,051 | ||||||
| Proceeds from Bounce Back Scheme Loan | - | 64,168 | ||||||
| Net Cash Provided By (Used In) Financing Activities | 25,411,919 | (8,733,927 | ) | |||||
The accompanying notes are an integral part of these consolidated financial statements.
F-7
180 LIFE SCIENCES CORP. AND SUBSIDIARIES
CONDENSED CONSOLIDATED STATEMENTS OF CASH FLOWS, continued
(Expressed in US Dollars)
| For the Years Ended | ||||||||
| December 31, | ||||||||
| 2021 | 2020 | |||||||
| Effect of Exchange Rate Changes on Cash | 75,473 | 140,311 | ||||||
| Net Increase In Cash | 6,115,964 | 2,025,147 | ||||||
| Cash - Beginning of Period | 2,108,544 | 83,397 | ||||||
| Cash - End of Period | $ | 8,224,508 | $ | 2,108,544 | ||||
| Supplemental Disclosures of Cash Flow Information: | ||||||||
| Cash paid during the period for income taxes | $ | - | $ | - | ||||
| Cash paid during the period for interest | $ | 35,351 | $ | - | ||||
| Non-cash investing and financing activities: | ||||||||
| Common stock issued upon conversion of KBL debt | $ | 1,931,388 | $ | - | ||||
| Common stock issued upon conversion of 180 debt | $ | 432,383 | $ | - | ||||
| Common stock issued in connection with repayment of related party loans and convertible notes | $ | 851,112 | $ | - | ||||
| Shares and warrants issued for Alpha Settlement | $ | 1,013,331 | $ | - | ||||
| Exchange of common stock equivalents for common stock | $ | 146 | $ | - | ||||
| Shares issued to settle accounts payable | $ | 1,750,000 | $ | - | ||||
| Reclassification of accrued issuable equity | $ | 43,095 | $ | - | ||||
| Recognition of beneficial conversion feature as loss on extinguishment of convertible note principal | $ | - | $ | 329,300 | ||||
| Redemption premium and restructuring fee recognized as an increase in convertible note principal | $ | - | $ | 557,436 | ||||
| Conversion of notes payable and accrued interest into common stock | $ | - | $ | 6,282,303 | ||||
| Deemed dividend - Conversion of Series A Convertible Preferred Stock into common stock | $ | - | $ | 4,349,035 | ||||
| Deemed dividend - Extinguishment on Series A Convertible Preferred stock | $ | - | $ | 565,659 | ||||
| Deemed dividend - Make-whole dividend on conversion of Series A Convertible preferred stock | $ | - | $ | 333,333 | ||||
| Deemed dividend - Beneficial conversion feature on Series A Convertible Preferred Stock | $ | - | $ | 223,710 | ||||
| Net non-cash liabilities assumed in Business Combination | $ | - | $ | 11,866,515 | ||||
| Financing of D&O insurance premium | $ | - | $ | 728,437 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
F-8
180 LIFE SCIENCES CORP. AND SUBSIDIARIES
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in US Dollars, except share amounts)
NOTE 1 - BUSINESS ORGANIZATION AND NATURE OF OPERATIONS
180 Life Sciences Corp., formerly known as KBL Merger Corp. IV (“180LS”, or together with its subsidiaries, the “Company”), was a blank check company organized under the laws of the State of Delaware on September 7, 2016. The Company was formed for the purpose of effecting a merger, share exchange, asset acquisition, share purchase, reorganization or similar business combination with one or more businesses.
180 Life Corp. (“180”, f/k/a 180 Life Sciences Corp. and CannBioRx Life Sciences Corp.) is a wholly-owned subsidiary of the Company and was incorporated in the State of Delaware on January 28, 2019. The Company is located in the United States (“U.S.”) and is a medical pharmaceutical company focused upon unmet medical needs in the areas of inflammatory diseases, fibrosis, and chronic pain by employing innovative research and, where appropriate, combination therapies, through 180’s three wholly-owned subsidiaries, 180 Therapeutics L.P. (“180 LP”), CannBioRex Pharmaceuticals Corp. (“CBR Pharma”), and Katexco Pharmaceuticals Corp. (“Katexco”). 180 LP, CBR Pharma and Katexco are together, the “180 Subsidiaries.” Katexco was incorporated on March 7, 2018 under the provisions of the British Corporation Act of British Columbia. Additionally, 180’s wholly-owned subsidiaries Katexco Callco, ULC, Katexco Purchaseco, ULC, CannBioRex Callco, ULC, and CannBioRex Purchaseco, ULC were formed in the Canadian Province of British Columbia on May 31, 2019 to facilitate the acquisition of Katexco, CBR Pharma and 180 LP. On July 1, 2021, the assets and liabilities of the Canadian companies (Katexco and CBR Pharma) were transferred to their respective subsidiaries, which are Katexco Pharmaceuticals Corp. (“Katexco U.S.”) and CannBioRex Pharma Limited (“CBR Pharma U.K.”).
The Company is a clinical stage biotechnology company focused on the development of therapeutics for unmet medical needs in chronic pain, inflammation, fibrosis and other inflammatory diseases, where anti-TNF therapy will provide a clear benefit to patients, by employing innovative research, and, where appropriate, combination therapy. We have three product development platforms:
| ● | fibrosis and anti-tumor necrosis factor (“TNF”); |
| ● | drugs which are derivatives of cannabidiol (“CBD”); and |
| ● | alpha 7 nicotinic acetylcholine receptor (“α7nAChR”). |
Reorganization and Business Combination
On July 16, 2019, 180 and each of 180 LP, Katexco and CBR Pharma completed a corporate restructuring, pursuant to which 180 LP, Katexco and CBR Pharma became wholly-owned subsidiaries of 180 (the “Reorganization”). It was determined that Katexco was the accounting acquirer in the Reorganization and the remaining companies were the accounting acquirees.
On November 6, 2020 (the “Closing Date”), the Company consummated the previously announced business combination (the “Business Combination”) following a special meeting of stockholders held on November 5, 2020, where the stockholders of the Company considered and approved, among other matters, a proposal to adopt that certain Business Combination Agreement (as amended, the “Business Combination Agreement”), dated as of July 25, 2019. Pursuant to the Business Combination Agreement, among other things, a subsidiary of the Company merged with and into 180, with 180 continuing as the surviving entity and a wholly- owned subsidiary of the Company (the “Merger”). The Merger became effective on November 6, 2020 (see Note 4 – Business Combination).
Risks and Uncertainties
Regarding the COVID-19 pandemic, a continuation or worsening of the levels of market disruption and volatility seen in the recent past could have an adverse effect on the Company’s ability to access capital, on the Company’s business, results of operations and financial condition. Management continues to monitor the developments and have taken active measures to protect the health of the Company’s employees, their families and the Company’s communities. The ultimate impact will depend heavily on the duration of the COVID-19 pandemic and public health responses, as well as the substance and pace of macroeconomic recovery, all of which are uncertain and difficult to predict considering the continuing evolving landscape of the COVID-19 pandemic and the public health responses to contain it.
F-9
Management has evaluated, and will continue to evaluate, the impact of the COVID-19 pandemic on the industry and has concluded that while it is reasonably possible that the virus could have a negative effect on the Company’s financial position, results of its operations and/or completion of business combination, the specific impact is not readily determinable as of the date of these consolidated financial statements. The follow up time for patient data and the statistical analysis for the Phase 2b Dupuytren’s Contracture clinical trial was delayed as a result of COVID-19, but such follow-up and statistical analysis are now completed and the Company announced the top-line data results from the Phase 2b trial on December 1, 2021. Additionally, COVID-19 has delayed the initiation of certain clinical trials and may delay the initiation of other clinical trials in the future or otherwise have a material adverse effect on our future operations. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
NOTE 2 - GOING CONCERN AND MANAGEMENT’S PLANS
The Company has not generated any revenues and has incurred significant losses since inception. For the year ended December 31, 2021, the Company incurred a net loss of $20,324,648 and used cash in operations of $19,371,428. As of December 31, 2021, the Company has an accumulated deficit of $68,682,286 and a working capital deficit of $8,498,193. The Company expects to invest a significant amount of capital to fund research and development. As a result, the Company expects that its operating expenses will increase significantly, and consequently will require significant revenues to become profitable. Even if the Company does become profitable, it may not be able to sustain or increase profitability on a quarterly or annual basis. The Company cannot predict when, if ever, it will be profitable. There can be no assurance that the intellectual property of the Company, or other technologies it may acquire, will meet applicable regulatory standards, obtain required regulatory approvals, be capable of being produced in commercial quantities at reasonable costs, or be successfully marketed. The Company plans to undertake additional laboratory studies with respect to the intellectual property, and there can be no assurance that the results from such studies or trials will result in a commercially viable product or will not identify unwanted side effects.
These consolidated financial statements have been prepared under the assumption of a going concern, which assumes that the Company will be able to realize its assets and discharge its liabilities in the normal course of business. The Company’s ability to continue its operations is dependent upon obtaining new financing for its ongoing operations. Future financing options available to the Company include equity financings and loans and if the Company is unable to obtain such additional financing timely, or on favorable terms, the Company may have to curtail its development, marketing and promotional activities, which would have a material adverse effect on its business, financial condition and results of operations, and it could ultimately be forced to discontinue its operations and liquidate. These matters raise substantial doubt about the Company’s ability to continue as a going concern for a reasonable period of time, which is defined as within one year after the date that the consolidated financial statements are issued. Realization of the Company’s assets may be substantially different from the carrying amounts presented in these consolidated financial statements and the accompanying consolidated financial statements do not include any adjustments that may become necessary, should the Company be unable to continue as a going concern.
NOTE 3 - SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Basis of Presentation
The accompanying consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the Unites States of America (“U.S. GAAP”). The Business Combination was accounted for as a reverse recapitalization, and 180 is deemed to be the accounting acquirer.
Consequently, the assets and liabilities and the historical operations that are reflected in these consolidated financial statements prior to the Business Combination are those of 180 Life Corp. and its subsidiaries. The preferred stock, common stock, additional paid in capital and earnings per share amount in these consolidated financial statements for the period prior to the Business Combination have been restated to reflect the recapitalization in accordance with the shares issued to the shareholders of the former parent, 180 Life Corp. as a result of the Business Combination.
F-10
Emerging Growth Company Disclosure Exemptions
The Company qualifies as an “emerging growth company,” as defined in the JOBS Act. For so long as the Company remains an emerging growth company, it is permitted and plans to rely on exemptions from certain disclosure requirements that are applicable to other public companies that are not emerging growth companies. These provisions include, but are not limited to: being permitted to have only two years of audited financial statements and only two years of related selected financial data and management’s discussion and analysis of financial condition and results of operations disclosure; an exemption from compliance with the auditor attestation requirement in the assessment of our internal control over financial reporting pursuant to Section 404 of the Sarbanes-Oxley Act; not being required to comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements; reduced disclosure obligations regarding executive compensation arrangements in our periodic reports, registration statements and proxy statements; and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. In addition, the JOBS Act permits emerging growth companies to take advantage of an extended transition period to comply with new or revised accounting standards applicable to public companies. The Company intends to take advantage of the exemptions discussed above.
Principles of Consolidation
On November 6, 2020 (the “Closing Date”), the Company consummated a business combination (the “Business Combination”) pursuant to which, among other things, a subsidiary of the Company merged with and into 180, with 180 continuing as the surviving entity and a wholly-owned subsidiary of the Company (the “Merger”, and the Company prior to the Merger sometimes referred to herein as “KBL”). The Business Combination was accounted for as a reverse recapitalization, and 180 is deemed to be the accounting acquirer. Consequently, the assets and liabilities and the historical operations that are reflected in these consolidated financial statements prior to the Business Combination are those of 180 Life Corp. and its subsidiaries. The preferred stock, common stock, additional paid in capital and earnings per share amount in these consolidated financial statements for the period prior to the Business Combination have been restated to reflect the recapitalization in accordance with the shares issued to the shareholders of the former parent, 180 Life Corp. as a result of the Business Combination.
The consolidated financial statements include the historical accounts of 180 Life Corp. as accounting acquirer along with its wholly-owned subsidiaries, and, effective with the closing of the Business Combination, 180LS as the accounting acquiree. All intercompany transactions and balances have been eliminated in consolidation.
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates, judgments, and assumptions that affect the reported amounts of assets, liabilities, revenues and expenses, together with amounts disclosed in the related notes to the consolidated financial statements. The Company’s significant estimates and assumptions used in these financial statements include, but are not limited to, the fair value of financial instruments warrants, options and equity shares; the valuation of stock-based compensation; and the estimates and assumptions related to impairment analysis of goodwill and other intangible assets long-lived assets. Certain of the Company’s estimates could be affected by external conditions, including those unique to the Company and general economic conditions. It is reasonably possible that these external factors could have an effect on the Company’s estimates and may cause actual results to differ from those estimates.
Accounting for Business Combinations
As required by U.S. GAAP, the Company records acquisitions under the acquisition method of accounting, under which the assets acquired and liabilities assumed are initially recorded at their respective fair values and any excess purchase price over the estimated fair value of net assets acquired is reflected as goodwill. The Company uses estimates and, in some instances, independent third-party valuation firms to assist in determining the fair values of assets acquired, liabilities assumed and contingent consideration, if any. Such estimates and valuations require significant assumptions, including projections of future events and operating performance. The estimated fair values are subject to change during the measurement period, which is limited to one year subsequent to the acquisition date.
F-11
Foreign Currency Translation
The Company’s reporting currency is the United States dollar. The functional currency of certain subsidiaries is the Canadian Dollar (“CAD”) or British Pound (“GBP”). Assets and liabilities are translated based on the exchange rates at the balance sheet date (0.7874 and 0.7847 for the CAD, 1.3510 and 1.3649 for the GBP as of December 31, 2021 and 2020, respectively), while expense accounts are translated at the weighted average exchange rate for the period (0.7977 and 0.7462 for the CAD and 1.3753 and 1.2843 for the GBP for the years ended December 31, 2021 and 2020, respectively). Equity accounts are translated at historical exchange rates. The resulting translation adjustments are recognized in stockholders’ equity as a component of accumulated other comprehensive income.
Comprehensive income is defined as the change in equity of an entity from all sources other than investments by owners or distributions to owners and includes foreign currency translation adjustments as described above. During the years ended December 31, 2021 and 2020, the Company recorded other comprehensive income of $180,133 and $484,083, respectively, as a result of foreign currency translation adjustments.
Foreign currency gains and losses resulting from transactions denominated in foreign currencies, including intercompany transactions, are included in results of operations. The Company recognized ($69) and $1,030 of foreign currency transaction (losses)/gains for the years ended December 31, 2021 and 2020, respectively. Such amounts have been classified within general and administrative expenses in the accompanying consolidated statements of operations and comprehensive loss.
Cash and Cash Equivalents
The Company considers all highly liquid investments with an original maturity of three months or less to be cash equivalents in the financial statements. The Company had no cash equivalents at December 31, 2021 or 2020. As of December 31, 2021, the Company had bank accounts in the United States and the United Kingdom. The Company’s cash deposits in United States and English financial institutions may at times may be in excess of the Federal Deposit Insurance Corporation (“FDIC”) or the Financial Services Compensation Scheme (“FSCS”) insurance limits, respectively. The Company has not experienced losses in such accounts and periodically evaluates the creditworthiness of its financial institutions.
Intangible Assets and In-Process Research and Development (“IPR&D”)
Intangible assets consist of licensed patents held by Katexco as well as technology licenses acquired in connection with the Reorganization. Licensed patents are amortized over the remaining life of the patent. Technology licenses represent the fair value of licenses acquired for the development and commercialization of certain licenses and knowledge. The technology licenses are amortized on a straight-line basis over the estimated useful lives of the underlying patents. It will be necessary to monitor and possibly adjust the useful lives of the licensed patents and technology licenses depending on the results of the Company’s research and development activities.
IPR&D assets represent the fair value assigned to technologies that were acquired on July 16, 2019 in connection with the Reorganization, which have not reached technological feasibility and have no alternative future use. IPR&D assets are considered to be indefinite-lived until the completion or abandonment of the associated research and development projects. During the period that the IPR&D assets are considered indefinite-lived, they are tested for impairment on an annual basis, or more frequently if the Company becomes aware of any events occurring or changes in circumstances that indicate that the fair value of the IPR&D assets are less than their carrying amounts. If and when development is complete, which generally occurs upon regulatory approval, and the Company is able to commercialize products associated with the IPR&D assets, these assets are then deemed definite-lived and are amortized based on their estimated useful lives at that point in time. If development is terminated or abandoned, the Company may record a full or partial impairment charge related to the IPR&D assets, calculated as the excess of the carrying value of the IPR&D assets over their estimated fair value.
Impairment of Long-Lived Assets
The Company reviews long-lived assets and certain identifiable assets for impairment whenever circumstances and situations change such that there is an indication that the carrying amounts may not be recovered. An impairment exists when the carrying value of the long-lived asset is not recoverable and exceeds its estimated fair value. No impairment charges were recorded during the years ended December 31, 2021 and 2020, respectively.
F-12
Goodwill
Goodwill represents the difference between the purchase price and the fair value of assets and liabilities acquired in a business combination. The Company reviews goodwill yearly, or more frequently whenever circumstances and situations change such that there is an indication that the carrying amounts may not be recovered, for impairment by initially considering qualitative factors to determine whether it is more likely than not that the fair value of a reporting unit is less than its carrying amount, including goodwill, as a basis for determining whether it is necessary to perform a quantitative analysis. If it is determined that it is more likely than not that the fair value of reporting unit is less than its carrying amount, a quantitative analysis is performed to identify goodwill impairment. If it is determined that it is not more likely than not that the fair value of the reporting unit is less than its carrying amount, it is unnecessary to perform a quantitative analysis. The Company may elect to bypass the qualitative assessment and proceed directly to performing a quantitative analysis. As of December 31, 2021, the Company elected to bypass the qualitative assessment and conducted a quantitative assessment whereby it was determined the fair value of the reporting unit (which the Company concluded was the consolidated entity), exceeded the carrying value and, accordingly, there was no impairment of goodwill.
Fair Value of Financial Instruments
The Company measures the fair value of financial assets and liabilities based on the guidance of Accounting Standards Codification (“ASC”) 820 “Fair Value Measurements” (“ASC 820”), which defines fair value, establishes a framework for measuring fair value, and expands disclosures about fair value measurements.
ASC 820 defines fair value as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. ASC 820 also establishes a fair value hierarchy, which requires an entity to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value. ASC 820 describes three levels of inputs that may be used to measure fair value:
| ● | Level 1 - Quoted prices in active markets for identical assets or liabilities; | |
| ● | Level 2 - Quoted prices for similar assets and liabilities in active markets or inputs that are observable; and | |
| ● | Level 3 - Inputs that are unobservable (for example, cash flow modeling inputs based on assumptions). |
The carrying amounts of certain of the Company’s financial instruments, consisting primarily of loans payable and convertible notes payable, approximate their fair values as presented in these consolidated financial statements due to the short-term nature of those instruments. The Company’s derivative liabilities were valued using level 3 inputs (see Note 9 – Derivative Liabilities for additional information).
Accrued Issuable Equity
The Company records accrued issuable equity when it is contractually obligated to issue shares and there has been a delay in the issuance of such shares. Accrued issuable equity is recorded and carried at fair value with changes in its fair value recognized in the Company’s consolidated statements of operations. Once the underlying shares of common stock are issued, the accrued issuable equity is reclassified as of the share issuance date at the then current fair market value of the common stock.
Stock-Based Compensation
The Company measures the cost of services received in exchange for an award of equity instruments based on the fair value of the award. The fair value of the award is measured on the grant date and is estimated by management based on observations of the recent cash sales prices of common stock. The fair value amount is then recognized over the period during which services are required to be provided in exchange for the award, usually the vesting period. Upon the exercise of an option or warrant, the Company issues new shares of common stock out of its authorized shares.
Derivative Liabilities and Convertible Instruments
The Company evaluates its debt and equity issuances to determine if those contracts or embedded components of those contracts qualify as derivatives requiring separate recognition in the Company’s financial statements. Entities must consider whether to classify contracts that may be settled in its own stock, such as warrants, as equity of the entity or as an asset or liability. If an event that is not within the entity’s control could require net cash settlement, then the contract should be classified as an asset or a liability rather than as equity.
F-13
The result of this accounting treatment is that the fair value of the embedded derivative is marked-to-market at each balance sheet date and recorded as a liability and the change in fair value is recorded in other (expense) income, net in the consolidated statements of operations. In circumstances where there are multiple embedded instruments that are required to be bifurcated, the bifurcated derivative instruments are accounted for as a single, compound derivative instrument. The classification of derivative instruments, including whether such instruments should be recorded as liabilities or as equity, is reassessed at the end of each reporting period. Equity instruments that are initially classified as equity that become subject to reclassification are reclassified to liability at the fair value of the instrument on the reclassification date. Derivative instrument liabilities are classified in the balance sheet as current or non-current based on whether or not net-cash settlement of the derivative instrument is expected within twelve months of the balance sheet date.
If the embedded conversion options do not require bifurcation, the Company then evaluates for the existence of a beneficial conversion feature by comparing the fair value of the Company’s underlying stock as of the commitment date to the effective conversion price of the instrument (the intrinsic value).
Debt discounts under these arrangements are amortized over the term of the related debt to their stated date of redemption and are classified in interest expense in the consolidated statements of operations. Preferred stock discounts are only accreted to their redemption value if redemption becomes probable.
Amendments to convertible instruments are evaluated as to whether they should be accounted for as a modification of the original instrument with no change to the accounting or, if the terms are substantially changed, as an extinguishment of the original instrument and the issuance of a new instrument.
The Company has computed the fair value of warrants, options, convertible notes and convertible preferred stock issued using the Monte-Carlo and Black-Scholes option pricing models. The expected term used for warrants, convertible notes and convertible preferred stock are the contractual life and the expected term used for options issued is the estimated period of time that options granted are expected to be outstanding. The Company utilizes the “simplified” method to develop an estimate of the expected term of “plain vanilla” option grants. The Company is utilizing an expected volatility figure based on a review of the historical volatilities, over a period of time, equivalent to the expected life of the instrument being valued, of similarly positioned public companies within its industry. The risk-free interest rate was determined from the implied yields from U.S. Treasury zero-coupon bonds with a remaining term consistent with the expected term of the instrument being valued.
Net Loss Per Common Share
Basic net loss per common share is computed by dividing net loss by the weighted average number of common shares outstanding during the period. Diluted net (loss) per common share is computed by dividing net (loss) by the weighted average number of common shares outstanding, plus the number of additional common shares that would have been outstanding if the common share equivalents had been issued (computed using the treasury stock or if converted method), if dilutive.
The following common share equivalents are excluded from the calculation of weighted average common shares outstanding, because their inclusion would have been anti-dilutive:
| For the Years Ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| Options | 2,741,000 | 50,000 | ||||||
| Warrants | 11,153,908 | 6,064,908 | ||||||
| Convertible debt (a) | - | 932,614 | ||||||
| Total potentially dilutive shares | 13,894,908 | 7,047,522 | ||||||
| a) | Represents shares issuable upon conversion of debt at various conversion prices, some of which were calculated using the fair value of the Company’s common stock at the respective balance sheet date. |
F-14
Research and Development
Research and development expenses are charged to operations as incurred. During the years ended December 31, 2021 and 2020, the Company incurred $1,000,769 and $2,217,371, respectively, of research and development expenses. As of December 31, 2021 and 2020, research and development expenses – related parties were $2,947,536 and $75,633, respectively. See Note 15 – Related Parties for more information on research and development expenses – related parties.
Income Taxes
The Company accounts for income taxes under the provisions of ASC Topic 740 “Income Taxes” (“ASC 740”).
The Company recognizes deferred tax assets and liabilities for the expected future tax consequences of items that have been included or excluded in the financial statements or tax returns. Deferred tax assets and liabilities are determined on the basis of the difference between the tax basis of assets and liabilities and their respective financial reporting amounts (“temporary differences”) at enacted tax rates in effect for the years in which the temporary differences are expected to reverse.
The Company utilizes a recognition threshold and measurement process for financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. The Company’s policy is to classify assessments, if any, for tax related interest as interest expense and penalties as general and administrative expenses in the consolidated statements of operations and comprehensive loss.
Recently Issued Accounting Pronouncements
Recently Adopted Accounting Pronouncements
In December 2019, the Financial Accounting Standards Board (the “FASB”) issued Accounting Standards Update (“ASU”) 2019-12, “Income Taxes (Topic 740): Simplifying the Accounting for Income Taxes,” which is intended to simplify various aspects related to accounting for income taxes. ASU 2019-12 removes certain exceptions to the general principles in Topic 740 and also clarifies and amends existing guidance to improve consistent application. The Company adopted ASU 2019-12 effective for January 1, 2021 and its adoption did not have a material impact on the Company’s consolidated financial statements and related disclosures.
Recently Issued But Not Yet Adopted Accounting Pronouncements
On May 3, 2021, FASB issued ASU 2021-04, Earnings Per Share (Topic 260), Debt—Modifications and Extinguishments (Subtopic 470-50), Compensation—Stock Compensation (Topic 718), and Derivatives and Hedging—Contracts in Entity’s Own Equity (Subtopic 815-40): Issuer’s Accounting for Certain Modifications or Exchanges of Freestanding Equity-Classified Written Call Options. This new standard provides clarification and reduces diversity in an issuer’s accounting for modifications or exchanges of freestanding equity-classified written call options (such as warrants) that remain equity classified after modification or exchange. This standard is effective for fiscal years beginning after December 15, 2021, including interim periods within those fiscal years. Issuers should apply the new standard prospectively to modifications or exchanges occurring after the effective date of the new standard. Early adoption is permitted, including adoption in an interim period. If an issuer elects to early adopt the new standard in an interim period, the guidance should be applied as of the beginning of the fiscal year that includes that interim period. The Company is evaluating this new standard.
F-15
NOTE 4 – BUSINESS COMBINATION
On November 6, 2020 (the “Closing Date”), the Company consummated the previously announced business combination (the “Business Combination”) following a special meeting of stockholders held on November 5, 2020, where the stockholders of the Company considered and approved, among other matters, a proposal to adopt that certain Business Combination Agreement (as amended, the “Business Combination Agreement”), dated as of July 25, 2019. References to “KBL” below refer to the Company prior to the Closing Date, then known as KBL Merger Corp. IV (“KBL”). Pursuant to the Business Combination Agreement, among other things, a subsidiary of the Company merged with and into 180, with 180 continuing as the surviving entity and a wholly-owned subsidiary of the Company (the “Merger”). The Business Combination was accounted for as a reverse recapitalization of 180. All of 180’s capital stock outstanding immediately prior to the merger was exchanged for (i) 15,736,348 shares of 180LS common stock, (ii) 2 shares of Class C and Class K Special Voting Shares exchangeable into 1,763,652 shares of 180LS common stock which are presented as outstanding in the accompanying Statement of Changes in Stockholders’ Equity due to the reverse recapitalization. KBL’s 6,928,645 outstanding shares of common stock are presented as being issued on the date of the Business Combination.
Below is a summary of the assets acquired and the liabilities assumed in connection with the Business Combination.
| Cash | $ | 3,006,235 | ||
| Prepaid expenses | 57,748 | |||
| Marketable securities held in Trust Account | 10,373,857 | |||
| Accounts payable and accrued expenses | (4,722,933 | ) | ||
| Convertible notes payable, net of debt discount | (2,504,045 | ) | ||
| Derivative liabilities (see Note 9) | (3,945,365 | ) | ||
| Due to/from Related Party | (201,859 | ) | ||
| Loans payable | (10,000 | ) | ||
| Promissory note with 180 | (496,161 | ) | ||
| Redemptions payable | (9,006,493 | ) | ||
| Net fair value of assets acquired and liabilities assumed | (7,449,016 | ) | ||
| Series A convertible preferred stock (see Note 13) | (1,411,265 | ) | ||
| Effect of reverse recapitalization | $ | (8,860,281 | ) |
Subsequent to the Business Combination, the marketable securities were released from the Trust Account, were converted into cash, and were used to settle the share redemption payable.
NOTE 5 - PREPAID EXPENSES AND OTHER CURRENT ASSETS
Prepaid expenses consist of the following as of December 31, 2021 and 2020:
| December 31, | ||||||||
| 2021 | 2020 | |||||||
| Insurance | $ | 2,151,487 | $ | 1,003,271 | ||||
| Research and development expense tax credit receivable | 644,513 | 409,470 | ||||||
| Professional fees | 80,783 | 104,080 | ||||||
| Value-added tax receivable | 24,411 | 37,751 | ||||||
| Taxes | 25,634 | 37,424 | ||||||
| Other | 49,755 | 14,418 | ||||||
| $ | 2,976,583 | $ | 1,606,414 | |||||
NOTE 6 – INTANGIBLE ASSETS
Intangible assets consist of the following as of December 31, 2021 and 2020:
| As of December 31, 2021 | As of December 31, 2020 | |||||||||||||||||||||||||||
| Remaining Amortization Period in Years at December 31, 2021 | Gross Asset Value | Accumulated Amortization | Net Carrying Value | Gross Asset Value | Accumulated Amortization | Net Carrying Value | ||||||||||||||||||||||
| Licensed patents | 14.6 | $ | 603,919 | $ | (110,759 | ) | $ | 493,160 | $ | 592,608 | $ | (76,766 | ) | $ | 515,842 | |||||||||||||
| Technology license | 17.5 | 1,658,550 | (202,797 | ) | 1,455,753 | 1,652,469 | (120,493 | ) | 1,531,976 | |||||||||||||||||||
| $ | 2,262,469 | $ | (313,556 | ) | $ | 1,948,913 | $ | 2,245,077 | $ | (197,259 | ) | $ | 2,047,818 | |||||||||||||||
F-16
Changes in the gross asset value of licensed patents and technology licenses from the dates acquired are the result of changes in the foreign currency exchange rate.
The Company recorded amortization expense of $116,297 and $116,841 during the years ended December 31, 2021 and 2020, respectively, related to intangible assets, which is included in general and administrative expense on the accompanying consolidated statements of operations and comprehensive loss.
Future amortization related to intangible assets is as follows:
| For the Years Ending December 31, | ||||
| 2022 | $ | 114,800 | ||
| 2023 | 114,800 | |||
| 2024 | 114,800 | |||
| 2025 | 114,800 | |||
| 2026 | 114,800 | |||
| Thereafter | 1,374,913 | |||
| $ | 1,948,913 | |||
NOTE 7 - ACCRUED EXPENSES
Accrued expenses consist of the following as of December 31, 2021 and 2020:
| December 31, | ||||||||
| 2021 | 2020 | |||||||
| Consulting fees | $ | 548,281 | $ | 1,718,559 | ||||
| Professional fees | 252,973 | 1,261,751 | ||||||
| Litigation accrual (1) | 300,000 | - | ||||||
| Employee and director compensation | 725,569 | 878,292 | ||||||
| Research and development fees | 91,737 | 17,817 | ||||||
| Interest | 25,433 | 184,576 | ||||||
| Other | 20,587 | 49,921 | ||||||
| $ | 1,964,580 | $ | 4,110,916 | |||||
| (1) | See Note 12 - Commitments and Contingencies, Potential Legal Matters. |
As of December 31, 2021 and 2020, accrued expenses - related parties were $18,370 and $454,951, respectively. See Note 15 - Related Parties for details.
NOTE 8 – ACCRUED ISSUABLE EQUITY
A summary of the accrued issuable equity activity during the years ended December 31, 2021 and 2020 is presented below:
| Balance at January 1, 2020 | $ | - | ||
| Additions | 43,095 | |||
| Balance at December 31, 2020 | 43,095 | |||
| Reclassification to equity | (43,095 | ) | ||
| Balance at December 31, 2021 | $ | - |
During the year ended December 31, 2020, the Company entered into a contractual arrangement for services in exchange for shares of common stock of the Company for fixed dollar amounts. Pursuant to the contractual agreement, the Company will issue an aggregate value of $5,000 common shares on a monthly basis and an aggregate of $30,000 of common shares at the end of each quarter. As of December 31, 2020, the Company recorded $43,095 of accrued issuable equity related to services. During the first quarter of 2021, this balance was reclassified to equity and as of December 31, 2021, there was no accrued issuable equity.
F-17
NOTE 9 - DERIVATIVE LIABILITIES
The following table sets forth a summary of the changes in the fair value of Level 3 derivative liabilities (except the Public Special Purpose Acquisition Companies (“SPAC”) warrants as defined below, which are Level 1 derivative liabilities) that are measured at fair value on a recurring basis:
| For the Year Ended December 31, 2021 | ||||||||||||||||||||||||
| Warrants | ||||||||||||||||||||||||
| Public | Private | Convertible | ||||||||||||||||||||||
| SPAC | SPAC | PIPE | Other | Notes | Total | |||||||||||||||||||
| Balance as of January 1, 2021 | $ | 3,795,000 | $ | 256,275 | $ | - | $ | 165,895 | $ | 225,800 | $ | 4,442,970 | ||||||||||||
| Extinguishment of derivative liabilities in connection with conversion of debt (1) | - | - | - | - | (591,203 | ) | (591,203 | ) | ||||||||||||||||
| Warrants issued in connection with the financing | - | - | 7,294,836 | - | - | 7,294,836 | ||||||||||||||||||
| Warrants issued relates to Alpha settlement (1) | - | - | - | 95,677 | - | 95,677 | ||||||||||||||||||
| Extinguishment of derivative liabilities in connection with the Alpha settlement (1) | - | - | - | - | (699,301 | ) | (699,301 | ) | ||||||||||||||||
| Change in fair value of derivative liabilities | 4,253,850 | 211,050 | (778,536 | ) | (73,680 | ) | 1,064,704 | 4,677,388 | ||||||||||||||||
| Balance as of December 31, 2021 | $ | 8,048,850 | $ | 467,325 | $ | 6,516,300 | $ | 187,892 | $ | - | $ | 15,220,367 | ||||||||||||
| (1) | See Note 11 – Convertible Notes Payable |
| For the Year Ended December 31, 2020 | ||||||||||||||||
| Warrants | Convertible Notes | Preferred Stock | Total | |||||||||||||
| Beginning balance as of January 1, 2020 | $ | - | $ | - | $ | - | $ | - | ||||||||
| Derivative liabilities assumed at date of Business Combination | 2,754,865 | $ | 23,500 | $ | 1,167,000 | $ | 3,945,365 | |||||||||
| Derecognition of derivative liabilities in connection with convertible note and preferred stock modification and exchanges | - | (723,336 | ) | (2,033,068 | ) | (2,756,404 | ) | |||||||||
| Issuance of derivative liabilities | - | 1,219,700 | 218,000 | 1,437,700 | ||||||||||||
| Change in fair value of derivative liabilities | 1,462,305 | (294,064 | ) | 648,068 | 1,816,309 | |||||||||||
| Ending balance as of December 31, 2020 | $ | 4,217,170 | $ | 225,800 | $ | - | $ | 4,442,970 | ||||||||
The fair value of the derivative liabilities as of December 31, 2021 were estimated using the Monte-Carlo and Black Scholes option pricing models, with the following assumptions used:
| December 31, 2021 | ||||
| Risk-free interest rate | 0.85% - 1.14 | % | ||
| Expected term in years | 2.59 - 4.15 | |||
| Expected volatility | 98.5 | % | ||
| Expected dividends | 0 | % | ||
In applying the Monte-Carlo and Black-Scholes option pricing models to derivatives assumed on November 6, 2020, the Company used the following assumptions:
| November 6, | ||||
| 2020 | ||||
| Risk-free interest rate | 0.08% - 0.40 | % | ||
| Expected term (years) | 0.26 - 5.01 | |||
| Expected volatility | 80% - 207 | % | ||
| Expected dividends | 0.00 | % | ||
In connection with the modification of certain convertible notes on November 25, 2020 (See Note 11 – Convertible Notes Payable for additional details), the Company applied the Monte-Carlo and Black-Scholes option pricing models to value embedded features as derivative liabilities with the following assumptions:
November 25, 2020 | ||||
| Risk-free interest rate | 0.06% - 0.09 | % | ||
| Expected term (years) | 0.24 - 0.54 | |||
| Expected volatility | 115% - 160 | % | ||
| Expected dividends | 0.00 | % | ||
F-18
SPAC Warrants
Public SPAC Warrants
Participants in KBL’s initial public offering received an aggregate of 11,500,000 Public SPAC Warrants (“Public SPAC Warrants”). Each Public SPAC Warrant entitles the holder to purchase one-half of one share of the Company’s common stock at an exercise price of $5.75 per half share ($11.50 per whole share), subject to adjustment. No fractional shares will be issued upon exercise of the Public Warrants. The Public Warrants are currently exercisable and will expire five years after the completion of the Business Combination or earlier upon redemption or liquidation. The Company may redeem the Public Warrants, in whole and not in part, at a price of $0.01 per Public Warrant upon 30 days’ notice (“30-day redemption period”), only in the event that the last sale price of the common stock equals or exceeds $18.00 per share for any 20 trading days within a 30-trading day period ending on the third trading day prior to the date on which notice of redemption is given, provided there is an effective registration statement with respect to the shares of common stock underlying such Public Warrants and a current prospectus relating to those shares of common stock is available throughout the 30-day redemption period. If the Company calls the Public Warrants for redemption as described above, the Company’s management will have the option to require all holders that wish to exercise Public Warrants to do so on a “cashless basis.” Management has determined that the Public Warrants contain a tender offer provision which could result in the Public Warrants settling for the tender offer consideration (including potentially cash) in a transaction that didn’t result in a change-in-control. This feature results in the Public Warrants being precluded from equity classification. Accordingly, the Public Warrants are classified as liabilities measured at fair value, with changes in fair value each period reported in earnings. The fair value of the Public SPAC Warrants on the date of the issuance was $1,978,000. At December 31, 2021 and 2020 the Public SPAC Warrants were revalued at $8,048,850 and $3,795,000, respectively, which resulted in $4,253,850 and $1,817,000 increase in the fair value of the derivative liabilities during the years ended December 31, 2021 and 2020, respectively and recorded in the accompanying consolidated statement of operations.
Private SPAC Warrants
Participants in KBL’s initial private placement received an aggregate of 502,500 Private SPAC Warrants (“Private SPAC Warrants”). Each Private Warrant entitles the holder to purchase one-half of one share of the Company’s common stock at an exercise price of $5.75 per half share ($11.50 per whole share), subject to adjustment. No fractional shares will be issued upon exercise of the warrants. The Private Warrants are currently exercisable and will expire five years after the completion of the Business Combination or earlier upon redemption or liquidation. The Private Warrants are non-redeemable so long as they are held by original holders or their permitted transferees. If the Private Warrants are held by other parties, the Company may redeem the Private Warrants, in whole and not in part, at a price of $0.01 per Warrant upon 30 days’ notice (“30-day redemption period”), only in the event that the last sale price of the common stock equals or exceeds $18.00 per share for any 20 trading days within a 30-trading day period ending on the third trading day prior to the date on which notice of redemption is given, provided there is an effective registration statement with respect to the shares of common stock underlying such Warrants and a current prospectus relating to those shares of common stock is available throughout the 30-day redemption period. If the Company calls the Private Warrants for redemption as described above, the Company’s management will have the option to require all holders that wish to exercise Private Warrants to do so on a “cashless basis.” Management has determined that the Private Warrants contain a tender offer provision which could result in the Private Warrants settling for the tender offer consideration (including potentially cash) in a transaction that didn’t result in a change-in-control. This feature (amongst others) results in the Private Warrants being precluded from equity classification. Accordingly, the Private Warrants are classified as liabilities measured at fair value, with changes in fair value each period reported in earnings. The fair value of the Private SPAC Warrants on the date of the issuance was $587,925. At December 31, 2021 and 2020 the Private SPAC Warrants were revalued at $467,325 and $256,275, respectively, which resulted in $211,250 increase and $331,650 decrease in the fair value of the derivative liabilities during the years ended December 31, 2021 and 2020, respectively. The increase and decrease in fair value of these derivative liabilities was recorded in the accompanying consolidated statement of operations.
PIPE Warrants
On February 23, 2021, the Company issued five-year warrants (the “PIPE Warrants”) to purchase 2,564,000 shares of common stock at an exercise price of $5.00 per share in connection with the private offering (see Note 13 – Stockholders’ Equity, Common Stock). The PIPE Warrants did not meet the requirements for equity classification due to the existence of a tender offer provision that could potentially result in cash settlement of the PIPE Warrants that didn’t meet the limited exception in the case of a change-in-control. Accordingly, the PIPE Warrants are liability-classified and the Company recorded the $7,294,836 fair value of the PIPE Warrants, which was determined using the Black-Scholes option pricing model, as derivative liabilities. The PIPE Warrants were revalued on December 31, 2021 at $6,516,300, which resulted in a $778,536 decrease in the fair value of the derivative liabilities during the year ended December 31, 2021.
The following assumptions were used to value the PIPE Warrants at issuance:
| February 23, 2021 | ||||
| Risk-free interest rate | 0.59 | % | ||
| Expected term in years | 5.00 | |||
| Expected volatility | 85 | % | ||
| Expected dividends | 0 | % | ||
F-19
Other Warrants
AGP Warrant
In connection with the closing of the Business Combination on November 6, 2020, the Company became obligated to assume five-year warrants for the purchase of 63,658 shares of the Company’s common stock at an exercise price of $5.28 per share (the “AGP Warrant Liability”) that had originally been issued by KBL to an investment banking firm in connection with a prior private placement.
On March 12, 2021, the Company issued a warrant to AGP (the “AGP Warrant”) to purchase up to an aggregate of 63,658 shares of the Company’s common stock at a purchase price of $5.28 per share, subject to adjustment, in full satisfaction of the existing AGP Warrant Liability. The exercise of the AGP Warrant is limited at any given time to prevent AGP from exceeding beneficial ownership of 4.99% of the then total number of issued and outstanding shares of the Company’s common stock upon such exercise. The warrant is exercisable at any time between May 2, 2021 and May 2, 2025. The newly issued AGP Warrant did not meet the requirements for equity classification due to the existence of a tender offer provision that could potentially result in cash settlement of the AGP Warrant that did not meet the limited exception in the case of a change-in-control. Accordingly, the AGP Warrant will continue to be liability-classified. The AGP Warrant was revalued on December 31, 2021 at $144,331 which resulted in a $21,564 decrease in the fair value of the derivative liabilities during the year ended December 31, 2021.
The following assumptions were used to value the AGP Warrant at issuance:
| March 12, 2021 | ||||
| Risk-free interest rate | 0.68 | % | ||
| Expected term in years | 3.84 | |||
| Expected volatility | 85 | % | ||
| Expected dividends | 0 | % | ||
Alpha Warrant
In connection with the Alpha Settlement Agreement (see Note 11 – Convertible Notes Payable) that was agreed to on July 29, 2021 (signed on July 31, 2021), the Company issued a three-year warrant for the purchase of 25,000 shares of the Company’s common stock at an exercise price of $7.07 per share (the “Alpha Warrant Liability” and the “Alpha Warrant”). The exercise of shares of the Alpha Warrant is limited at any given time to prevent Alpha from exceeding a beneficial ownership of 4.99% of the then total number of issued and outstanding shares of the Company’s common stock upon such exercise. The warrant is exercisable until August 2, 2024. The newly issued Alpha Warrant did not meet the requirements for equity classification due to the existence of a tender offer provision that could potentially result in cash settlement of the Alpha Warrant that did not meet the limited exception in the case of a change-in-control. Accordingly, the Alpha Warrant is liability-classified and the Company recorded the $95,677 fair value of the Alpha Warrant, which was determined using the Black-Scholes option pricing model, as a derivative liability. The Alpha Warrant was revalued on December 31, 2021 at $43,561, which resulted in a $52,116 decrease in the fair value of the derivative liabilities during the year ended December 31, 2021.
The following assumptions were used to value the Alpha Warrant at issuance:
| July 29, 2021 | ||||
| Risk-free interest rate | 0.37 | % | ||
| Expected term in years | 3.00 | |||
| Expected volatility | 85 | % | ||
| Expected dividends | 0 | % | ||
Convertible Notes
The convertible notes issued in 2020 had embedded features that were bifurcated and recorded as derivative liabilities. Between January 15, 2021 and February 5, 2021, the fair value of derivative liabilities extinguished in connection with the conversion of debt (see Note 11 – Convertible Notes Payable) was estimated using the Monte-Carlo and Black Scholes option pricing models with the following assumptions used:
| January 15, 2021 to | ||||
| February 5, 2021 | ||||
| Risk-free interest rate | 0.00% - 0.14 | % | ||
| Expected term in years | 0.02 - 0.18 | |||
| Expected volatility | 120% - 161 | % | ||
| Expected dividends | 0 | % | ||
F-20
At the end of the second quarter of 2021, the Alpha Capital Note (see Note 11 – Convertible Notes Payable) that was the only convertible note with an outstanding balance and the full amount of the July 31, 2021 Alpha Settlement Agreement was accrued as of that date. On July 31, 2021, the Company recorded the extinguishment of the Alpha Capital Note, the related derivative liabilities and the balance of the settlement accrual. See Note 11 - Convertible Notes Payable for additional details.
Warrant Activity
A summary of the warrant activity (including the August 2021 PIPE Warrants which are equity-classified; see Note 13 - Stockholders’ Equity) during the years ended December 31, 2021 and 2020 is presented below:
| Number
of Warrants |
Weighted Average Exercise Price | Weighted Average Remaining Life in Years | Intrinsic Value | |||||||||||
| Outstanding, January 1, 2020 | - | - | ||||||||||||
| Issued | 6,064,908 | 11.43 | ||||||||||||
| Outstanding, December 31, 2020 | 6,064,908 | 11.43 | ||||||||||||
| Issued | 5,089,000 | 6.24 | ||||||||||||
| Exercised | - | - | ||||||||||||
| Cancelled | - | - | ||||||||||||
| Expired | - | - | ||||||||||||
| Outstanding, December 31, 2021 | 11,153,908 | $ | 9.06 | 4.1 | ||||||||||
| Exercisable, December 31, 2021 | 11,153,908 | $ | 9.06 | 4.1 | ||||||||||
A summary of outstanding and exercisable warrants as of December 31, 2021 is presented below:
| Warrants Outstanding | Warrants Exercisable | |||||||||||||
| Weighted | ||||||||||||||
| Average | ||||||||||||||
| Exercise | Number of | Remaining | Number of | |||||||||||
| Price | Shares | Life in Years | Shares | |||||||||||
| $ | 5.00 | 2,564,000 | 4.2 | 2,564,000 | ||||||||||
| $ | 5.28 | 63,658 | 3.3 | 63,658 | ||||||||||
| $ | 7.07 | 25,000 | 2.6 | 25,000 | ||||||||||
| $ | 7.50 | 2,500,000 | 4.6 | 2,500,000 | ||||||||||
| $ | 11.50 | 6,001,250 | 3.9 | 6,001,250 | ||||||||||
| 11,153,908 | 4.1 | 11,153,908 | ||||||||||||
A summary of outstanding and exercisable warrants as of December 31, 2020 is presented below:
| Warrants Outstanding | Warrants Exercisable | |||||||||||||
| Weighted | ||||||||||||||
| Average | ||||||||||||||
| Exercise | Number of | Remaining | Number of | |||||||||||
| Price | Shares | Life in Years | Shares | |||||||||||
| $ | 11.50 | 6,001,250 | 4.9 | 6,001,250 | ||||||||||
| 5.28 | 63,658 | - | ||||||||||||
| 6,064,908 | 4.9 | 6,001,250 | ||||||||||||
F-21
NOTE 10 - LOANS PAYABLE
The following tables summarize the activity of loans payable during the years ended December 31, 2021 and 2020:
| Principal Balance at January 1, 2021 | Forgiveness/ Adjusted to Other Income | Principal Repaid in Cash | New Issuances | Effect of Foreign Exchange Rates | Principal Balance at December 31, 2021 | |||||||||||||||||||
| Kingsbrook | $ | 150,000 | $ | - | $ | (150,000 | ) | $ | - | $ | - | $ | - | |||||||||||
| Paycheck Protection Program | 53,051 | (11,670 | ) | (69 | ) | - | - | 41,312 | ||||||||||||||||
| Bounce Back Loan Scheme | 68,245 | - | (4,724 | ) | - | (2,352 | ) | 61,169 | ||||||||||||||||
| First Assurance Funding | 655,593 | - | (655,593 | ) | 1,618,443 | - | 1,618,443 | |||||||||||||||||
| Other loans payable | 155,320 | - | - | - | - | 155,320 | ||||||||||||||||||
| Total loans payable | 1,082,209 | $ | (11,670 | ) | $ | (810,386 | ) | $ | 1,618,443 | $ | (2,352 | ) | 1,876,244 | |||||||||||
| Less: loans payable - current portion | 968,446 | 1,828,079 | ||||||||||||||||||||||
| Loans payable - non-current portion | $ | 113,763 | $ | 48,165 | ||||||||||||||||||||
| Principal Balance at January 1, 2020 | Forgiveness/ Adjusted to Other Income | Principal Repaid in Cash | New Issuances | Effect of Foreign Exchange Rates | Principal Balance at December 31, 2020 | |||||||||||||||||||
| Kingsbrook | $ | - | $ | - | $ | - | $ | 150,000 | $ | - | $ | 150,000 | ||||||||||||
| Paycheck Protection Program | - | - | - | 53,051 | - | 53,051 | ||||||||||||||||||
| Bounce Back Loan Scheme | - | - | - | 68,245 | - | 68,245 | ||||||||||||||||||
| First Assurance Funding | - | - | (347,222 | ) | 1,002,815 | - | 655,593 | |||||||||||||||||
| Other loans payable | 116,250 | - | - | 39,070 | - | 155,320 | ||||||||||||||||||
| Total loans payable | 116,250 | $ | - | $ | (347,222 | ) | $ | 1,313,181 | $ | - | 1,082,209 | |||||||||||||
| Less: loans payable - current portion | 116,250 | 968,446 | ||||||||||||||||||||||
| Loans payable - non-current portion | $ | - | $ | 113,763 | ||||||||||||||||||||
Loans Payable, Current Portion
| Simple Interest Rate | December 31, | December 31, | ||||||||||
| Loan payable issued September 18, 2019 | 8 | % | $ | 50,000 | $ | 50,000 | ||||||
| Loan payable issued October 29, 2019 | 8 | % | 69,250 | 69,250 | ||||||||
| Loan payable issued February 5, 2020 | 8 | % | 3,500 | 3,500 | ||||||||
| Loan payable issued March 31, 2020 | 8 | % | 4,537 | 4,537 | ||||||||
| Loan payable issued June 8, 2020 | 8 | % | 5,000 | 5,000 | ||||||||
| Loan payable issued June 8, 2020 | 8 | % | 5,000 | 5,000 | ||||||||
| Kingsbrook loan issued June 12, 2020 | 8 | % | - | 150,000 | ||||||||
| Loan payable issued July 15, 2020 * | 8 | % | 4,695 | 4,695 | ||||||||
| Loan payable issued October 13, 2020 | 8 | % | 13,337 | 13,337 | ||||||||
| Loan payable issued December 10, 2020 | 8 | % | - | 655,594 | ||||||||
| Current portion of PPP Loans (1) | 1 | % | 41,312 | 7,533 | ||||||||
| Current portion of Bounce Back Loans (1) | 1 | % | 13,005 | - | ||||||||
| Loan payable issued December 10, 2021 | 2 | % | 1,618,443 | - | ||||||||
| $ | 1,828,079 | $ | 968,446 | |||||||||
| * | These loans are denominated in currencies other than USD. |
| (1) | See Loans Payable, Non-Current Portion for a description of the PPP Loans and the Bounce Back Loans. |
F-22
Loans Payable, Non-Current Portion
The non-current portion of the Company’s loans payable as of December 31, 2021 and 2020 are as follows:
| Simple Interest Rate | December 31, 2021 |
December 31, 2020 |
Maturity Date | |||||||||||
| PPP loan payable issued May 5, 2020 | 1.0 | % | 41,312 | $ | 51,051 | 5/4/2022 | ||||||||
| PPP loan payable issued April 24, 2020 | 1.0 | % | - | 2,000 | 4/23/2022 | |||||||||
| BBLS loan payable issued June 10, 2020 | 2.5 | % | 61,170 | 68,245 | 6/10/2026 | |||||||||
| Subtotal | 102,482 | 121,296 | ||||||||||||
| Less: Current portions of BBLS/PPP loans, respectively (see above) | (54,317 | ) | (7,533 | ) | ||||||||||
| Non-current portion | $ | 48,165 | $ | 113,763 | ||||||||||
During April and May 2020, the Company received loans in the aggregate amount of $53,051 (the “PPP Loans”), under the Payroll Protection Program (“PPP”), to support continuing employment during the COVID-19 pandemic.
Effective March 27, 2020, legislation referred to as the Coronavirus Aid, Relief, and Economic Security Act (the “CARES Act”) was passed to benefit companies in the U.S. that were significantly impacted by the pandemic. Under the terms of the CARES Act, as amended by the Paycheck Protection Program Flexibility Act of 2020, the Company is eligible to apply for and receive forgiveness for all or a portion of their respective PPP Loans. Such forgiveness will be determined, subject to limitations, based on the use of the loan proceeds for certain permissible purposes as set forth in the PPP, including, but not limited to, payroll costs (as defined under the PPP) and mortgage interest, rent or utility costs (collectively, “Qualifying Expenses”) incurred during the 24 weeks subsequent to funding, and on the maintenance of employee and compensation levels, as defined, following the funding of the PPP Loan. The Company intends to use the proceeds of their PPP Loans for Qualifying Expenses. However, no assurance is provided that the Company will be able to obtain forgiveness of the PPP Loans in whole or in part. Any amounts not forgiven incur interest at 1.0% per annum and monthly repayments of principal and interest are deferred for six months after the date of disbursement. While the Company’s PPP loans currently have a two-year maturity, the amended law will permit the Company to request a five-year maturity. As of December 31, 2021 and 2020, the Company recorded accrued interest of $163 and $354, respectively, related to the PPP loans. During the years ended December 31, 2021 and 2020, the Company recorded interest expense of $1,636 and $354, respectively, related to the PPP loans.
On May 19, 2021, the Company applied for loan forgiveness for the amount of $51,051 in connection with amounts borrowed by Katexco under the Paycheck Protection Program. On August 5, 2021, the Company was notified that $9,670 was forgiven in connection with the PPP loan. The Company has appealed the decision and requested the full amount of the PPP loan be forgiven.
On September 30, 2021, the Company adjusted a portion of the PPP loan in the amount of $2,000 to other income since such amount was a grant to 180LS by the government, and it did not need to be repaid.
On June 10, 2020, the Company received GBP £50,000 (USD $64,353) of cash proceeds pursuant to the Bounce Back Loan Scheme (“BBLS”), which provides financial support to businesses across the UK that are losing revenue, and seeing their cashflow disrupted, as a result of the COVID-19 outbreak. The BBLS is unsecured and bears interest at 2.5% per annum. The maximum loan amount is GBP £50,000 and the length of the loan is six years, with payments beginning 12 months after the date of disbursement. Early repayment is allowed, without early repayment fees. As of December 31, 2021 and 2020, the Company recorded accrued interest of GBP £778 (USD $1,051) and GBP £514 (USD $702), respectively, related to the BBLS loan. During the years ended December 31, 2021 and 2020, the Company recorded interest expense of GBP £778 (USD $1,051) and GBP£514 (USD $702), respectively, related the BBLS loan.
On June 12, 2020, the Company entered into a promissory note agreement with Kingsbrook Opportunities Master Fund LP for an aggregate principal sum of $150,000, which bears interest at 15% per annum and matures on August 31, 2021. On March 3, 2021, the Company repaid the Kingsbrook loans payable in cash for an aggregate of $162,452, which included the principal amount of $150,000 and accrued interest of $12,452.
During the year ended December 31, 2021, the Company paid an aggregate of $655,593 in full satisfaction of the 2020 directors and officers insurance policy and $4,724 in partial satisfaction of the Bounce Back Loan Scheme.
On December 10, 2021, the Company entered into a financing arrangement for a Directors and Officers Insurance Policy (the “D&O Insurance”) with First Assurance Funding to finance $1,618,443 of a total D&O Insurance amount of $2,005,502 inclusive of premiums, taxes, and fees. As of December 31, 2021, a total of $1,618,443 remains financed in loans payable, due in monthly installments of $161,844.
F-23
Loans Payable – Related Parties
Loans payable to related parties (the “Related Party Loans”) consist of loans payable to certain of the Company’s officers, directors and a greater than 10% stockholder. The Company had the following loans payable to related parties outstanding as of December 31, 2021 and 2020:
Simple Interest Rate | December 31, | December 31, | ||||||||||
| Loan payable issued September 18, 2019 | 8 | % | $ | 50,000 | $ | 50,000 | ||||||
| Loan payable issued October 8, 2019 | 0 | % | 4,000 | 4,000 | ||||||||
| Loan payable issued October 20, 2019 * | 8 | % | - | 81,463 | ||||||||
| Loan payable issued October 28, 2019 * | 8 | % | - | 7,088 | ||||||||
| Loan payable issued October 29, 2019 | 8 | % | - | 40,000 | ||||||||
| Loan payable issued October 29, 2019 | 8 | % | - | 10,000 | ||||||||
| Loan payable issued November 27, 2019 * | 8 | % | - | 20,515 | ||||||||
| Loan payable issued December 11, 2019 | 8 | % | - | 10,342 | ||||||||
| Loan payable issued January 14, 2020 | 8 | % | - | 4,726 | ||||||||
| Loan payable issued January 20, 2020 | 8 | % | - | 137,382 | ||||||||
| Loan payable issued January 30, 2020 * | 8 | % | - | 7,088 | ||||||||
| Loan payable issued February 5, 2020 | 8 | % | 3,500 | 3,500 | ||||||||
| Loan payable issued February 28, 2020 * | 8 | % | - | 19,261 | ||||||||
| Loan payable issued March 31, 2020 | 8 | % | 4,537 | 4,537 | ||||||||
| Loan payable issued April 2, 2020 | 8 | % | - | 1,871 | ||||||||
| Loan payable issued April 2, 2020 | 8 | % | - | 1,564 | ||||||||
| Loan payable issued April 13, 2020 | 8 | % | - | 12,875 | ||||||||
| Loan payable issued April 13, 2020 | 8 | % | - | 12,905 | ||||||||
| Loan payable issued April 27, 2020 * | 8 | % | - | 7,962 | ||||||||
| Loan payable issued May 19, 2020 | 8 | % | - | 2,152 | ||||||||
| Loan payable issued May 30, 2020 * | 8 | % | - | 7,962 | ||||||||
| Loan payable issued May 30, 2020 | 8 | % | - | 7,890 | ||||||||
| Loan payable issued June 17, 2020 | 8 | % | 485 | 485 | ||||||||
| Loan payable issued July 15, 2020 | 8 | % | 5,503 | 5,503 | ||||||||
| Loan payable issued August 25, 2020 * | 8 | % | - | 9,162 | ||||||||
| Loan payable issued October 8, 2020 * | 8 | % | 8,708 | 8,796 | ||||||||
| Loan payable issued October 15, 2020 | 8 | % | - | 10,094 | ||||||||
| Loan payable issued October 14, 2020 * | 8 | % | 4,544 | 4,544 | ||||||||
| Loan payable issued October 1, 2020 * | 8 | % | - | 10,253 | ||||||||
| Loan payable issued November 4, 2020 * | 8 | % | - | 9,162 | ||||||||
| $ | 81,277 | $ | 513,082 | |||||||||
| * | These loans are denominated in currencies other than USD. |
At issuance, the Related Party Loans provided for a maturity date upon the earliest of (a) the consummation of the Business Combination; (b) June 30, 2020; or (c) 60 days after the respective issuance date. On July 1, 2020, the Company amended the terms of the Related Party Loans to extend the maturity terms to the earlier of (a) the closing of a qualified financing; or (b) November 1, 2020. The terms of all loan extensions were reviewed and were deemed to be modifications, rather than extinguishments.
On February 10, 2021, the Company entered into amended loan agreements to modify the terms of certain loan agreements in the aggregate principal amount of $432,699, previously entered into with Sir Marc Feldmann and Dr. Lawrence Steinman, the Co-Executive Chairmen of the Board of Directors. The loan agreements were extended and modified to be paid back at the Company’s discretion, either by 1) repayment in cash, or 2) by converting the outstanding amounts into shares of common stock at the same price per share as the next financing transaction. Subsequently, on February 25, 2021, and effective as of the date of the original February 10, 2021 amendments, the Company determined that such amendments were entered into in error and each of Sir Feldmann and Dr. Steinman rescinded such February 10, 2021 amendments pursuant to their entry into Confirmations of Rescission acknowledgements. As such, the amendments to allow Sir Feldmann and Dr. Steinman the option to convert such loans into shares of common stock were never effective.
On April 12, 2021, the Company entered into amended loan agreements with Sir Marc Feldmann and Dr. Lawrence Steinman, the Co-Executive Chairman of the Board of Directors, which extended the maturity date of all of their outstanding loan agreements to September 30, 2021.
On that day, they elected to exchange an aggregate principal of $433,374 and aggregate accrued interest of $61,530 into an aggregate of 82,484 shares of the Company’s common stock at a price of $6.00 per share, pursuant to the terms of the agreement (see Note 13 - Stockholders’ Equity).
F-24
Interest Expense on Loans Payable
For the year ended December 31, 2021, the Company recognized interest expense and interest expense — related parties associated with outstanding loans, of $24,019 and $38,874, respectively.
For the year ended December 31, 2020, the Company recognized interest expense and interest expense — related parties associated with outstanding loans, of $23,709 and $35,973, respectively.
As of December 31, 2021, the Company had accrued interest and accrued interest — related parties associated with outstanding loans, of $24,212 and $812, respectively. See Note 15 — Related Parties for additional details.
As of December 31, 2020, the Company had accrued interest and accrued interest — related parties associated with outstanding loans, of $24,824 and $37,539, respectively. See Note 15 — Related Parties for additional details.
NOTE 11 - CONVERTIBLE NOTES PAYABLE
The table below details the convertible notes payable activity during the years ended December 31, 2021 and 2020:
| Maturity | |||||||||||||||||||||||||||||
| Date | |||||||||||||||||||||||||||||
| (as amended, | 01/01/21 | Impact | 12/31/21 | ||||||||||||||||||||||||||
| Effective | if | Principal | of | Conversions | Common | Principal | |||||||||||||||||||||||
| Date | applicable) | Balance | Extinguishment | to Common Stock | Shares Issued | Balance | |||||||||||||||||||||||
| Dominion | 06/12/20 | 02/11/21 | $ | 833,334 | $ | - | $ | (833,334) | 338,393 | $ | - | ||||||||||||||||||
| Kingsbrook | 06/12/20 | 02/11/21 | 101,000 | - | (101,000) | 33,770 | - | ||||||||||||||||||||||
| Alpha Capital | 06/12/20 | 02/11/21 | 616,111 | (316,111) | (300,000) | 94,960 | - | ||||||||||||||||||||||
| Bridge Note | 12/27/19 | 08/28/21 | 365,750 | - | (365,750) | 158,383 | - | ||||||||||||||||||||||
| Total | $ | 1,916,195 | $ | (316,111) | $ | (1,600,084) | 625,506 | $ | - | ||||||||||||||||||||
| For The Year Ended December 31, 2020 | ||||||||||||||||||||||||||||||||||||
| Maturity | ||||||||||||||||||||||||||||||||||||
| Date | ||||||||||||||||||||||||||||||||||||
| (as amended, | 01/01/20 | Amortization | Conversions | 12/31/20 | ||||||||||||||||||||||||||||||||
| Effective | if | Principal | of Debt | Impact of | to Common | Principal | ||||||||||||||||||||||||||||||
| Date | applicable) | Balance | Debt Issued | Debt Discount | Discount | Extinguishment | Stock | Balance | ||||||||||||||||||||||||||||
| Dominion | 06/12/20 | 02/11/21 | $ | - | $ | 1,805,556 | $ | (722,966 | ) | $ | 134,134 | $ | 588,832 | $ | (972,222 | ) | $ | 833,334 | ||||||||||||||||||
| Kingsbrook | 06/12/20 | 02/11/21 | - | 1,796,411 | (685,615 | ) | 127,227 | 558,388 | (1,695,411 | ) | 101,000 | |||||||||||||||||||||||||
| Alpha Capital | 06/12/20 | 02/11/21 | - | 1,111,111 | (800,421 | ) | 94,786 | 705,635 | (495,000 | ) | 616,111 | |||||||||||||||||||||||||
| Amended Senior Note | 07/25/19 | 08/28/21 | 1,405,695 | - | - | - | - | - | - | |||||||||||||||||||||||||||
| Amended Senior Note (1 | 07/25/19 | 08/28/21 | 1,081,251 | - | - | - | - | (1,768,779 | ) | - | ||||||||||||||||||||||||||
| Bridge Note | 12/27/19 | 08/28/21 | 250,000 | - | - | - | - | - | 275,000 | |||||||||||||||||||||||||||
| Bridge Note | 01/03/20 | 08/28/21 | - | 82,500 | - | - | - | - | 90,750 | |||||||||||||||||||||||||||
| Total | $ | 2,736,946 | $ | 4,795,578 | $ | (2,209,002 | ) | $ | 356,147 | $ | 1,852,855 | $ | (4,931,412 | ) | $ | 1,916,195 | ||||||||||||||||||||
| [1] | See Note 10 - Convertible Notes Payable - Extinguishment of Senior Note and Issuance of New Note for additional details. |
| [2] | See Note 10 - Convertible Notes Payable - Bridge Notes for additional details. |
| [3] | See Note 10 - Convertible Notes Payable - Amended Bridge Notes for additional details. |
F-25
The following table details the convertible notes payable – related parties activities during the years ended December 31, 2021 and 2020:
| For the Year Ended December 31, 2021 | ||||||||||||||||||||||||||||
| Effective Date | Maturity Date (as amended, if applicable) | 01/01/21 Principal Balance | Debt Issued | Unpaid Interest Capitalized to Principal | Settlement Debt | Conversions to Common Stock | 12/31/21 Principal Balance | |||||||||||||||||||||
| 180 LP Convertible Note | 09/24/13 | 09/25/15 | 160,000 | - | - | - | (160,000 | ) | - | |||||||||||||||||||
| 180 LP Convertible Note | 06/16/14 | 06/16/17 | 10,000 | - | - | (10,000 | ) | - | - | |||||||||||||||||||
| 180 LP Convertible Note | 07/08/14 | 07/08/17 | 100,000 | - | - | - | (100,000 | ) | - | |||||||||||||||||||
| Total | $ | 270,000 | $ | - | $ | - | $ | (10,000 | ) | $ | (260,000 | ) | $ | - | ||||||||||||||
| For the year ended December 31, 2020 | ||||||||||||||||||||||||||||
| Effective Date | Maturity Date (as amended, if applicable) | 01/01/20 Principal Balance | Debt Issued | Unpaid Interest Capitalized to Principal | Amendment to Senior Notes | Conversions to Common Stock | 12/31/20 Principal Balance | |||||||||||||||||||||
| Amended Senior Notes (1) | 07/25/19 | 08/28/21 | $ | 184,604 | $ | - | $ | 34,760 | $ | 51,396 | (270,760 | ) | $ | - | ||||||||||||||
| 180 LP Convertible Note | 09/24/13 | 09/25/15 | 160,000 | - | - | - | - | 160,000 | ||||||||||||||||||||
| 180 LP Convertible Note | 06/16/14 | 06/16/17 | 10,000 | - | - | - | - | 10,000 | ||||||||||||||||||||
| 180 LP Convertible Note | 07/08/14 | 07/08/17 | 100,000 | - | - | - | - | 100,000 | ||||||||||||||||||||
| Total | $ | 454,604 | $ | - | $ | 34,760 | $ | 51,396 | $ | (270,760 | ) | $ | 270,000 | |||||||||||||||
Dominion, Kingsbrook and Alpha Convertible Promissory Note
Upon closing of the Business Combination, the Dominion (defined below), Kingsbrook and Alpha (defined below) Convertible Promissory Notes were assumed.
Dominion Convertible Promissory Notes
| Dominion | ||||||||||||
| Principal | Debt Discount | Net | ||||||||||
| Balance at January 1, 2020 | $ | - | $ | - | $ | - | ||||||
| Assumption of Note | 1,805,556 | - | 1,805,556 | |||||||||
| Debt discount at assumption | - | (722,966 | ) | (722,966 | ) | |||||||
| Amortization of debt discount | - | 134,134 | 134,134 | |||||||||
| Impact of extinguishment | - | 588,832 | 588,832 | |||||||||
| Impact of conversion | (972,222 | ) | - | (972,222 | ) | |||||||
| Balance at December 31, 2020 | $ | 833,334 | $ | - | $ | 833,334 | ||||||
| Impact of conversion | (833,334 | ) | - | (833,334 | ) | |||||||
| Balance at December 31, 2021 | $ | - | $ | - | $ | - | ||||||
On June 12, 2020 (the “Dominion Issue Date”), KBL entered into a $1,666,667 10% Secured Convertible Promissory Note and $138,889 10% Senior Secured Convertible Extension Promissory Note (together the “Dominion Convertible Promissory Notes”) with Dominion Capital LLC (“Dominion”), which was issued to Dominion in conjunction with 400,000 shares of common stock (the “Dominion Commitment Shares”). In conjunction with the transaction, KBL entered into a series of Leak Out Agreements in which certain parities agreed that they would not sell, dispose or otherwise transfer, in aggregate more than 5% of the composite daily trading volume of the common stock of KBL. Pursuant to the Leak-Out Agreement between the KBL and Caravel CAD Fund Ltd., KBL issued 404,245 restricted shares of common stock (“Leak-Out Shares”).
F-26
The Dominion Convertible Promissory Notes had a debt discount due to original issue discount, third-party fees directly attributed to the issuance, leak-out shares, a derivative liability, a beneficial conversion feature and warrants. The debt discount assumed at the Business Combination for this note was $722,996, which has been amortized to interest expense over the term of the debt. See Note 9 – Derivative Liabilities for more information on the derivative liabilities related to this note.
The Company has agreed to pay the principal amount, together with interest at the annual rate of 10% (unless the Company defaults, which increases the interest rate to 15%) (including 10% guaranteed interest), with principal and accrued interest on the Dominion Convertible Promissory Notes due and payable on February 11, 2021 (the “Dominion Maturity Date”), unless converted under terms and provisions as set forth within the Dominion Convertible Promissory Notes. The Dominion Convertible Promissory Notes provided Dominion with the right to convert, at any time, all or any part of the outstanding principal and accrued but unpaid interest into shares of the Company’s common stock at a conversion price of $5.28 per share. The Dominion Convertible Promissory Notes required the Company to reserve at least 868,056 and 114,584 shares of common stock from its authorized and unissued common stock to provide for all issuances of common stock under the 10% Secured Convertible Promissory Note and 10% Senior Secured Convertible Extension Promissory Note, respectively. However, the Dominion Convertible Promissory Notes provided that the aggregate number of shares of common stock issued to the Dominion under the Dominion Convertible Promissory Notes shall not exceed 4.99% of the total number of shares of common stock outstanding as of the closing date unless the Company obtains stockholder approval of the issuance (the “the Beneficial Ownership Limitation”). Dominion, upon not less than sixty-one (61) days’ prior notice to the Company, may increase or decrease the Beneficial Ownership Limitation; provided, that the Beneficial Ownership Limitation in no event exceeds 9.99% of the number of shares of the common stock outstanding immediately after giving effect to the issuance of shares of common stock upon conversion of the Dominion Convertible Promissory Notes held by Dominion.
During the year ended December 31, 2021, the Company recorded interest expense of $31,080 as of December 31, 2021 associated with the Dominion Convertible Promissory Notes.
During the year ended December 31, 2020, the Company recorded interest expense and amortization of debt discount of $77,067 and $134,164, respectively, and accrued interest of $52,254 as of December 31, 2020 associated with the Dominion Convertible Promissory Notes.
See Convertible Debt Conversions of the Dominion, Kingsbrook and Alpha Convertible Promissory Notes further on in this note for the details related to the 2020 and 2021 conversions of the notes.
Kingsbrook Convertible Promissory Note
| Kingsbrook | ||||||||||||
| Principal | Debt Discount | Net | ||||||||||
| Balance at January 1, 2020 | $ | - | $ | - | $ | - | ||||||
| Assumption of Note | 1,796,411 | - | 1,796,411 | |||||||||
| Debt discount at assumption | - | (685,615 | ) | (685,615 | ) | |||||||
| Amortization of debt discount | - | 127,227 | 127,227 | |||||||||
| Impact of extinguishment | - | 558,388 | 558,388 | |||||||||
| Impact of conversion | (1,695,411 | ) | - | (1,695,411 | ) | |||||||
| Balance at December 31, 2020 | $ | 101,000 | $ | - | $ | 101,000 | ||||||
| Impact of conversion | (101,000 | ) | - | (101,000 | ) | |||||||
| Balance at December 31, 2021 | $ | - | $ | - | $ | - | ||||||
On June 12, 2020 (the “Kingsbrook Issue Date”), KBL entered into a $1,657,522 10% Secured Convertible Promissory Note and $138,889 10% Senior Secured Convertible Extension Promissory Note (together with “Kingsbrook Convertible Promissory Notes”) with Kingsbrook Opportunities Master Fund LP (“Kingsbrook”), which was issued to Kingsbrook in conjunction with 250,000 shares of common stock (the “Kingsbrook Commitment Shares”).
F-27
The Kingsbrook Convertible Promissory Notes had a debt discount due to original issue discount, third-party fees directly attributed to the issuance, a derivative liability, a beneficial conversion feature and warrants. The debt discount assumed at the Business Combination for this note was $685,615, which has been amortized to interest expense over the term of the debt. See Note 9 – Derivative Liabilities for more information on the derivative liabilities related to this note.
The Company has agreed to pay the principal amount, together with guaranteed interest at the annual rate of 10% (unless the Company defaults, which increases the interest rate to 15%), with principal and accrued interest on the Kingsbrook Convertible Promissory Notes due and payable on February 11, 2021 (the “Maturity Date”), unless converted under terms and provisions as set forth within the Kingsbrook Convertible Promissory Notes. The Kingsbrook Convertible Promissory Notes provide Kingsbrook with the right to convert, at any time, all or any part of the outstanding principal and accrued but unpaid interest into shares of the Company’s common stock at a conversion price of $5.28 per share. The Kingsbrook Convertible Promissory Notes require the Company to reserve at least 1,823,275 and 114,584 shares of common stock from its authorized and unissued common stock to provide for all issuances of common stock under the 10% Secured Convertible Promissory Note and 10% Senior Secured Convertible Extension Promissory Note, respectively. However, the Kingsbrook Convertible Promissory Notes provide that the aggregate number of shares of common stock issued to Kingsbrook under the Kingsbrook Convertible Promissory Notes shall not exceed 4.99% of the total number of shares of common stock outstanding as of the closing date unless the Company has obtained stockholder approval of the issuance (the “the Beneficial Ownership Limitation”). Kingsbrook, upon not less than sixty-one (61) days’ prior notice to the Company, may increase or decrease the Beneficial Ownership Limitation; provided, that the Beneficial Ownership Limitation in no event exceeds 9.99% of the number of shares of the common stock outstanding immediately after giving effect to the issuance of shares of common stock upon conversion of the Kingsbrook Convertible Promissory Notes held by Kingsbrook.
During the year ended December 31, 2021, the Company recorded interest expense of $10,010 as of December 31, 2021 associated with the Kingsbrook Convertible Promissory Notes.
During the year ended December 31, 2020, the Company recorded interest expense and amortization of debt discount of $61,315 and $127,228, respectively, and accrued interest of $0 as of December 31, 2020 associated with the Kingsbrook Convertible Promissory Notes.
See Convertible Debt Conversions of the Dominion, Kingsbrook and Alpha Convertible Promissory Notes further on in this note for the details related to the 2020 and 2021 conversions of the notes.
Alpha Convertible Promissory Note
| Alpha | ||||||||||||
| Principal | Debt Discount | Net | ||||||||||
| Balance at January 1, 2020 | $ | - | $ | - | $ | - | ||||||
| Assumption of Note | 1,111,111 | - | 1,111,111 | |||||||||
| Debt discount at assumption | - | (800,421 | ) | (800,421 | ) | |||||||
| Amortization of debt discount | - | 94,786 | 94,786 | |||||||||
| Impact of extinguishment | - | 705,635 | 705,635 | |||||||||
| Impact of conversion | (495,000 | ) | - | (495,000 | ) | |||||||
| Balance at December 31, 2020 | $ | 616,111 | $ | - | $ | 616,111 | ||||||
| Impact of extinguishment | (316,111 | ) | - | (316,111 | ) | |||||||
| Impact of conversion | (300,000 | ) | - | (300,000 | ) | |||||||
| Balance at December 31, 2021 | $ | - | $ | - | $ | - | ||||||
On September 8, 2020 (the “Alpha Issue Date”), KBL entered into a $1,111,111 10% Secured Convertible Promissory Note (the “Alpha Convertible Promissory Note”) with Alpha Capital Anstalt (“Alpha”), which was issued to the Holder in conjunction with 100,000 shares of common stock (the “Alpha Capital Anstalt Commitment Shares”).
F-28
The Alpha Convertible Promissory Notes had a debt discount due to original issue discount, third-party fees directly attributed to the issuance, a derivative liability, a beneficial conversion feature and warrants. The debt discount assumed at the Business Combination for this note was $800,421, which has been amortized to interest expense over the term of the debt. See Note 9 – Derivative Liabilities for more information on the derivative liabilities related to this note.
The Company has promised to pay the principal amount, together with guaranteed interest at the annual rate of 10% (unless the Company defaults, which increases the interest rate to 15%), with principal and accrued interest on the Alpha Convertible Promissory Note due and payable on April 7, 2021 (the “Maturity Date”), unless converted under terms and provisions as set forth within the Alpha Capital Anstalt Convertible Note. The Alpha Convertible Promissory Note provides Alpha with the right to convert, at any time, all or any part of the outstanding principal and accrued but unpaid interest into shares of the Company’s common stock at a conversion price of $5.28 per share. The Alpha Convertible Promissory Note provides that the aggregate number of shares of common stock issued to Alpha under the Alpha Convertible Promissory Note shall not exceed 4.99% of the total number of shares of common stock outstanding as of the closing date unless the Company has obtained stockholder approval of the issuance (the “the Beneficial Ownership Limitation”). Alpha, upon not less than sixty-one (61) days’ prior notice to the Company, may increase or decrease the Beneficial Ownership Limitation; provided, that the Beneficial Ownership Limitation in no event exceeds 9.99% of the number of shares of the common stock outstanding immediately after giving effect to the issuance of shares of common stock upon conversion of the Alpha Convertible Promissory Note held by Alpha.
During the year ended December 31, 2021, the Company recorded interest expense of $58,510 as of December 31, 2021 associated with the Alpha Convertible Promissory Notes.
During the year ended December 31, 2020, the Company recorded interest expense and amortization of debt discount of $28,962 and $94,787, respectively, and accrued interest of $47,504 as of December 31, 2020 associated with the Alpha Convertible Promissory Notes.
See Convertible Debt Conversions of the Dominion, Kingsbrook and Alpha Convertible Promissory Notes further on in this note for the details related to the 2020 and 2021 conversions of the notes.
2020 Extinguishment of the Dominion, Kingsbrook and Alpha Convertible Promissory Notes
On November 25, 2020, the Company entered into an amended agreement with Dominion, and Alpha to amend the secured convertible promissory notes in the original aggregate principal amount of $4,713,078 (after giving effect to a 10% original issue discount) that the Company issued pursuant to a purchase agreement (the “Notes”) so that the fixed conversion price of the Notes, during the 90 day period following November 6, 2020, shall be equal to the lower of: (A) ninety-six percent (96%) of the lowest volume weighted average price of the common stock of the Company on the NASDAQ Capital Market during the five trading day period ending on the trading day immediately prior to the applicable conversion date and (B) $5.28; provided, that in no event shall the fixed conversion price be lower than $2.00 (in each case, as appropriately adjusted for any stock dividend, stock split, stock combination, reclassification or similar transaction that proportionately decreases or increases the number of shares of common stock prior to such date). No other changes were made to the Notes as a result of the amendment agreement. The change of the conversion price of the Notes, triggered the most-favored-nation clause and changed the conversion price of the Series A Convertible Preferred Stock to be the same price as the Notes.
The Company determined that while the cash flows of the Secured Convertible Notes did not change upon the amendment (maturity and interest rate remained the same), the increase of the fair value of the conversion feature exceeded 10% of the carrying value of the Secured Convertible Notes prior to the amendment, and accordingly, the amendment should be accounted for as an extinguishment. In recording the extinguishment, the Company compared the reacquisition price of the post-amended Secured Convertible Notes in the aggregate amount of $5,932,778 to the net carrying value of the derivative liability and pre-amended Secured Convertible Notes in the aggregate amount of $2,880,524. As a result, the Company recorded a loss on extinguishment in the aggregate amount of $3,052,254.
F-29
2020 Convertible Debt Conversions of the Dominion, Kingsbrook and Alpha Convertible Promissory Notes
The holders of the Secured Convertible Promissory Notes elected to convert principal and interest into shares of the Company’s common stock during 2020 as follows:
| Loss on | ||||||||||||||||||||||||||||
| Fair Value | Extinguishment | |||||||||||||||||||||||||||
| Principal | Derivative | Total | Common | of | of | |||||||||||||||||||||||
| Balance | Interest | Liabilities | Amount | Shares | Shares | Convertible | ||||||||||||||||||||||
| Converted | Converted | Converted | Converted | Issued | Issued | Notes | ||||||||||||||||||||||
| Dominion Convertible Promissory Note | $ | 972,222 | $ | 97,222 | $ | 201,216 | $ | 1,270,660 | 464,287 | $ | 1,275,525 | $ | 4,865 | |||||||||||||||
| Kingsbrook Convertible Promissory Note | 1,695,411 | 169,541 | 378,335 | $ | 2,243,287 | 816,769 | 2,198,155 | (45,132 | ) | |||||||||||||||||||
| Alpha Capital Convertible Promissory Note | 495,000 | 12,528 | 123,485 | $ | 631,013 | 238,572 | 691,304 | 60,291 | ||||||||||||||||||||
| Total | $ | 3,162,633 | $ | 279,291 | $ | 703,036 | $ | 4,144,960 | 1,519,628 | $ | 4,164,984 | $ | 20,024 | |||||||||||||||
After the closing of the Business Combination, from November 27, 2020 to December 31, 2020, the holders of the Company’s convertible promissory notes converted an aggregate of $3,441,924, which includes accrued interest of $279,291, which is owed under such convertible notes into an aggregate of 1,519,628 shares of our common stock, pursuant to the terms of such notes, as amended, at conversion prices of between $2.00 and $2.31 per share.
Default of Certain Convertible Promissory Notes
On December 31, 2020, the Company filed a Current Report on Form 8-K with the SEC which disclosed, among other things, that the condensed consolidated financial statements of the Company, which were prepared based on the information and representations received from the former KBL management, for the interim period ended September 30, 2020, should no longer be relied upon due to errors in the condensed consolidated financial statements and should be restated. As a result, the Company recognized a derivative liability related to an arguable default of the Secured Convertible Notes, which was valued using a Monte Carlo Simulation.
2021 Convertible Debt Conversion/Extinguishment of the Dominion, Kingsbrook and Alpha Convertible Promissory Notes
The holders of the Secured Convertible Promissory Notes elected to convert principal and interest into shares of the Company’s common stock during 2021 as follows:
| Loss on | ||||||||||||||||||||||||||||
| Fair Value | Extinguishment | |||||||||||||||||||||||||||
| Principal | Derivative | Total | Common | of | of | |||||||||||||||||||||||
| Balance | Interest | Liabilities | Amount | Shares | Shares | Convertible | ||||||||||||||||||||||
| Converted | Converted | Converted | Converted | Issued | Issued | Notes | ||||||||||||||||||||||
| Dominion Convertible Promissory Note | $ | 833,333 | $ | 83,333 | $ | 133,033 | $ | 1,049,700 | 338,393 | $ | 1,255,037 | $ | (205,337 | ) | ||||||||||||||
| Kingsbrook Convertible Promissory Note | 101,000 | 10,100 | 136,800 | 247,900 | 33,770 | 174,253 | 73,647 | |||||||||||||||||||||
| Alpha Capital Convertible Promissory Note | 300,000 | 12,417 | 321,370 | 633,787 | 94,960 | 511,834 | 121,953 | |||||||||||||||||||||
| Total | $ | 1,234,333 | $ | 105,850 | $ | 591,203 | $ | 1,931,387 | 467,123 | $ | 1,941,124 | $ | (9,737 | ) | ||||||||||||||
During the third quarter of 2021, certain noteholders elected to convert certain convertible notes payable with an aggregate principal balance of $1,234,333 and an aggregate accrued interest balance of $105,850 into an aggregate of 467,123 shares of the Company’s common stock at conversion prices ranging from $2.45-$3.29 per share. The shares issued upon the conversion of the convertible promissory notes had a fair value at issuance of $1,941,124.
F-30
Alpha – Extinguishment
On February 3, 2021, an event of default was triggered under a convertible note held by Alpha Capital Anstalt (“Alpha” and the “Alpha Capital Note”), which resulted in an increase in the fair value of the bifurcated derivative liability (the default provision) associated with the remaining principal of the Alpha Capital Note.
On July 29, 2021, the Company reached a settlement agreement with Alpha (the “Alpha Settlement Agreement”), which was signed on July 31, 2021, which provided for Alpha to convert the remaining principal and accrued interest associated with the convertible note in exchange for 150,000 shares of the Company’s common stock plus a three-year warrant to purchase 25,000 additional shares of the Company’s common stock at an exercise price of $7.07 per share. The Company determined that the shares and warrants had an aggregate value of $1,156,177 as of July 29, 2021, which exceeded the aggregate $1,109,008 carrying value of the combined principal, accrued interest and derivative liability associated with the Alpha Capital Note as of July 29, 2021. Because the settlement amount provides additional information about a situation that existed as of July 29, 2021, the Company recorded an accrual as of June 30, 2021 for the $47,169 difference between the value of the securities offered in settlement and the carrying value of the liabilities, which was reflected within (loss) gain on settlement of liabilities in the accompanying condensed consolidated statements of operations. On July 29, 2021, the $1,156,177 aggregate carrying value of the principal, accrued interest, derivative liability and settlement accrual associated with the Alpha Capital Note were extinguished while the $1,060,500 fair value of the common stock was recorded within equity and the $95,677 fair value of the Alpha Warrant was recorded as a derivative liability (see Note 9, Derivative Liabilities for additional information).
Senior Notes
On July 25, 2019, the Company issued Senior Secured Notes (the “Senior Notes”) totaling $1,200,000 of which an aggregate of $175,000 was issued to the former Chief Executive Officer and a director of the Company. The Senior Notes bear interest at a rate of 15% per annum and matured on November 15, 2019. Any accrued and unpaid interest portion is capitalized to principal on a monthly basis. Pursuant to the terms of the Senior Notes, the maturity date may be extended an additional 30 days at the option of the Company if the Securities and Exchange Commission’s review of the documents filed in connection with the Business Combination has taken more than 30 days. In the event of an event of default: a) the Company is required to notify the holders of these notes (the “Holders”) within one business day of any such occurrence; b) the interest rate increases to 18% per annum; and c) the Holder may require the Company to redeem any or all of the outstanding principal and interest together with a 25% premium.
The Senior Notes rank senior to all outstanding and future indebtedness of the Company and its subsidiaries and are secured by: a) the Company’s equity interests in its subsidiaries; b) guarantees issued by those subsidiaries; and c) assets of those subsidiaries.
The Senior Notes, plus accrued and unpaid interest, and any outstanding late charges, automatically convert into common shares immediately prior to the occurrence of the Business Combination at the conversion price of $4.23 per share. If the Company issues any shares of its common stock or securities that are effectively common stock equivalents prior to the Business Combination at a price of less than $4.23 per share, then the conversion price per share will be adjusted so that the Holders receive the same conversion price. The above represents a contingent beneficial conversion feature that will be accounted for when the contingency is resolved.
F-31
On January 13, 2020, the Company and holders of a series of Senior Secured Notes (the “Senior Notes”) agreed to exchange the Senior Notes for new Senior Secured Notes (the “Amended Senior Notes”) with amended terms (the “Senior Note Amendments”). Pursuant to the Amended Senior Notes, the note holders waived all events of default associated with the Senior Notes and the aggregate principal amount and accrued interest of $1,282,205 and $6,411, respectively, was converted to principal in the aggregate amount of $1,846,052 (consisting of $1,282,205 of the outstanding principal of the Senior Notes, $6,411 of accrued interest reclassified to principal, $200,000 of restructuring fees and $357,436 of redemption premiums), of which $186,988 and $935, of aggregate principal and accrued interest, respectively, owed to the former Chief Executive Officer and a director of the Company, was converted to principal in the aggregate amount of $239,320 (consisting of $186,988 of the outstanding principal of the Senior Notes, $935 of accrued interest reclassified to principal and $51,396 of redemption premiums). See above in Note 11 – Convertible Notes Payable for a table displaying the impact of the increase in the principal under the column titled Amendment to Senior Note and Bridge Notes.
The Company accounted for the amendment to the Senior Notes as note extinguishments, since the present value of future cash flows under the Amended Senior Notes was substantially different than the future cash flows under the Senior Notes. Accordingly, the Company recognized a loss on extinguishment of $886,736, consisting of the issuance of the Amended Senior Note in the aggregate principal amount of $1,846,052, partially offset by the derecognition of the aggregate carrying amount of the extinguished Senior Notes of $1,288,616, plus the immediately recognized beneficial conversion feature of $329,300 arising from the modified conversion terms of the Amended Senior Notes.
The Amended Senior Notes rank senior to all outstanding and future indebtedness of the Company and its subsidiaries and are secured by: a) the Company’s equity interests in its subsidiaries; b) guarantees issued by those subsidiaries; and c) assets of those subsidiaries.
The Amended Senior Notes were convertible into common stock of the Company at any time following issuance until maturity and automatically convert into common stock of the Company immediately prior to the occurrence of the Business Combination, in either event, at a conversion price of $4.23 per share. If the Company issues any shares of its common stock, or securities that are effectively common stock equivalents, prior to the Business Combination at a price of less than $4.23 per share, then the conversion price per share would be adjusted to the price at which those common shares (or equivalents) were issued.
The Amended Senior Notes bear interest at a rate of 15% per annum and matured in February 2020. On June 12, 2020, the Company entered into an additional amendment with each noteholder to extend the maturity dates from February 2020 to August 2021. Unpaid interest is reclassified to the principal on a monthly basis.
In the event of default: a) the Company is required to notify the holders of these notes within one business day of any such occurrence; b) the interest rate increases to 18% per annum; and c) the holder may require the Company to redeem any or all of the outstanding principal and interest together with a 25% premium.
Additional Amendment to an Amended Senior Note
On June 12, 2020, the Company, KBL, and the holder of an Amended Senior Note in the aggregate principal amount of $1,661,136 agreed that (i) such Amended Senior Note will automatically convert into 404,265 shares of the Company’s common stock upon the Business Combination, and (ii) the holder of such Amended Senior Note and its affiliates shall not sell or dispose more than 5% of the daily trading volume of such shares of common stock as reported by Bloomberg, LP.
Extinguishment of Senior Note and Issuance of New Note
On June 12, 2020, the Company, KBL, certain investors (the “Purchasers”) and the holder (the “Initial Purchaser”) of an Amended Senior Note in the aggregate principal and interest amount of $1,528,360 (consisting of principal of $1,510,113 and accrued interest payable of $18,247) entered into a Securities Purchase Agreement pursuant to which (i) the Amended Senior Note was extinguished, and (ii) KBL sold to the Purchasers a secured promissory note which is secured by the intellectual property of the Company. Such transaction closed on June 29, 2020. See above in Note 11 – Convertible Notes Payable for a table displaying the impact of extinguishing the aforementioned $1,510,113 of principal under the column titled Amendment to Senior Note and Bridge Notes. Concurrent with the transaction, on June 12, 2020, the Company, KBL, the Purchasers and Kingsbrook entered into a guaranty agreement pursuant to which the Company is a guarantor to the notes issued by KBL to the Purchasers and Kingsbrook. As of September 30, 2020, the Company determined that contingent payments under the guaranty agreement were not probable.
Additionally, in connection with the Securities Purchase Agreement, the Company issued the Initial Purchaser a non-convertible loan payable in the principal amount of $150,000 which bears interest at a rate of 15% per annum, payable at maturity. The note matures on August 31, 2021 (see Note 10 - Loans Payable). On March 3, 2021, the Company repaid this loan in cash for an aggregate $162,452, which included the principal of $150,000 and accrued interest of $12,452.
F-32
Conversion of Senior Notes at Close of Business Combination
On November 6, 2020, upon the consummation of the Business Combination, the Company issued 482,894 shares of common stock, par value $0.0001, to the holders of the Senior Notes, as a result of the automatic conversion of promissory notes in the principal amount of about $2,039,539 and accrued interest of $77,779, or an aggregate of $2,117,318, as per the closing of the Merger pursuant to the Business Combination Agreement, dated as of July 25, 2019, by and among the Company, KBL Merger Sub, Inc., 180 Life Corp., Katexco Pharmaceuticals Corp., CannBioRex Pharmaceuticals Corp., 180 Therapeutics L.P. and Lawrence Pemble in his capacity as stockholder representative.
Bridge Notes
On January 3, 2020 and December 27, 2019, the Company issued convertible bridge notes in the aggregate amount of $82,500 and $250,000 under the same terms. The total outstanding principal amount of convertible bridge notes of $332,500 (the “Bridge Notes”) and the respective accrued interest will automatically convert into a portion of the 17.5 million shares of KBL common stock to be received upon the consummation of the Business Combination Agreement at a conversion price equal to the lesser of $6.00 per KBL share or 60% of the implied valuation at such time, as defined. The Bridge Notes accrue interest at 15% per annum. The contingently adjustable, non-bifurcated beneficial conversion feature associated with the convertible note will be accounted for at the time the contingency is resolved. The Bridge Notes matured on June 30, 2020. The Company may elect to prepay the Bridge Notes at any time without penalty, however, the holder may elect to receive shares of common stock of the Company in lieu of prepayment at the holder’s discretion. The Company analyzed the embedded conversion option of the convertible note at issuance and determined the embedded conversion option contains a contingent beneficial conversion feature that will be accounted for when the contingency is resolved. See below for further details regarding the amendment of the Bridge Notes and the conversion terms.
Amended Bridge Notes
On July 7, 2020, effective June 29, 2020, the Company entered into an amendment agreement with each Bridge Noteholder (the “Amended Bridge Notes”). Pursuant to the terms of the Amended Bridge Notes, the principal under each Amended Bridge Note is increased by 10% and the Amended Bridge Notes mature upon the earlier of (i) the date that the Registration Statement, which refers to the Form S-4 Registration Statement filed with the Commission by KBL, relating to the Business Combination, including the exchange of shares of common stock of the Company for shares of common stock of KBL, which is declared effective by the SEC; (ii) such date in which all amounts due and owing under the Amended Bridge Notes become due and payable pursuant to the terms of the agreement; and August 28, 2021 (“Maturity Date”). See above in Note 11 – Convertible Notes Payable for a table displaying the impact of the increase in the 10% of principal under the column titled Amendment to Senior Note and Bridge Notes. The Amended Bridge Notes can be converted at the following options:
| ● | at any time prior to the Maturity Date, at the option of the holder, the remaining outstanding principal amount of the Amended Bridge Notes, and any accrued but unpaid interest, may be converted into shares of common stock; or |
| ● | automatically at the Maturity Date, the remaining outstanding principal amounts of these Amended Bridge Notes and any accrued but unpaid interest, will automatically convert (“Automatic Conversion”). |
Depending on the timing of the conversion, the holder will receive either:
| ● | shares of common stock of the Company, if the Business Combination has not occurred prior to such Maturity Date; or |
| ● | shares of KBL if the Business Combination has occurred prior to such Maturity Date. |
In either case above, the number of conversion shares equal to (A) the outstanding principal amount plus interest being converted, divided by (B) the lesser of (i) $4.23 per share or (ii) the per share price equal to 0.60 multiplied by the per share price of one share of common stock sold by the Company as part of a PIPE transaction (or the deemed value of one share of common stock as reasonably determined by the board of directors of the Company sold in the PIPE transaction if the securities sold in the PIPE transaction include units or other securities convertible into shares of common stock), subject to equitable adjustment for any stock splits, combinations or similar events affecting the Company. As per the agreement, a PIPE transaction is defined as a private placement or public offering of shares of common stock of KBL for purposes of raising additional capital to fund the Business Combination or other matters. The Company determined that the embedded features of the Amended Bridge Notes were (i) a fixed price conversion option which represented a beneficial conversion feature of de minimis value and (ii) a redemption feature with de minimis value.
F-33
Second Amendment to the Bridge Notes
On October 7, 2020, the Company entered into an additional amendment with each Amended Bridge Noteholder pursuant to which the Amended Bridge Notes will no longer mature upon the date that the Registration Statement is declared effective by the SEC. Since the change in cash flows was not more than 10%, this amendment was deemed to be a modification.
Conversions of Bridge Notes
On March 8, 2021, the holders of the Company’s convertible bridge notes, which were issued in December 27, 2019 and January 3, 2020 to various purchasers, converted an aggregate of $432,384, which included accrued interest of $66,633 owed under such convertible bridge notes, into an aggregate of 158,383 shares of common stock pursuant to the terms of such notes, as amended, at a conversion price of $2.73 per share.
180 LP Convertible Notes
In connection with the Reorganization, the Company assumed $270,000 of debt related to convertible notes payable (the “Notes”), of which $10,000 is owed to the former Chief Executive Officer of 180 LP and $260,000 is owed to a founder and director of the Company.
Principal of $160,000 due under the Notes accrues interest at a rate of 5.0% per annum and principal of $110,000, accrues interest at 2.5% per annum. Interest is compounded annually. Effective upon the closing of the first issuance of convertible preferred units (or units with similar rights) with proceeds of at least $1,000,000 (the “Qualified Financing”), all of the outstanding principal and interest under these Notes will automatically be converted into other equity interests of the Company of the same class issued to other investors in the Qualified Financing, at a conversion price equal to 80% of the price per unit of the Qualified Financing securities paid by the other investors. The Notes contain contingent beneficial conversion features, which will be accounted for at the time the conversion price is known, and the contingency is resolved.
During the second quarter of 2021, the Company repaid a certain related party convertible note payable in cash for the principal amount of $10,000 and $1,873 of accrued interest. During the third quarter of 2021, the $260,000 remaining principal balance of convertible notes payable owed to a related party, plus $96,208 of related accrued interest, was converted into 59,368 shares of the Company’s common stock, pursuant to a debt conversion agreement dated September 30, 2021.
Interest on Convertible Notes
During the years ended December 31, 2021 and 2020, the Company recorded interest expense of $109,767 and $915,371, respectively, related to convertible notes payable, and recorded interest expense - related parties of $42,529 and $32,452, respectively, related to convertible notes payable - related parties. During the years ended December 31, 2021 and 2020, the Company capitalized an aggregate of $0 and $228,099 of accrued interest, respectively, and $0 and $34,760 of accrued interest – related parties, respectively, to note principal.
As of December 31, 2021 and 2020, accrued interest expense related to convertible notes payable was $0 and $182,181, respectively, and accrued interest expense - related parties related to convertible notes payable - related parties was $0 and $124,833, respectively, which is included in accrued expenses and accrued expenses - related parties, respectively, on the accompanying consolidated balance sheets.
NOTE 12 - COMMITMENTS AND CONTINGENCIES
Litigation and Other Loss Contingencies
The Company records liabilities for loss contingencies arising from claims, assessments, litigation, fines, penalties and other sources when it is probable that a liability has been incurred and the amount of the loss can be reasonably estimated. The Company has no liabilities recorded for loss contingencies as of December 31, 2021. See Potential Legal Matters – Action Against Former Executives of KBL and Cantor Fitzgerald & Co. Breach of Contract below for information related to a December 31, 2021 accrual.
Potential Legal Matters
Action Against Former Executive of KBL
On September 1, 2021, the Company initiated legal action in the Chancery Court of Delaware against Dr. Marlene Krauss ("Dr. Krauss") and two of her affiliated companies, KBL IV Sponsor, LLC and KBL Healthcare Management, Inc. (collectively, the “KBL Affiliates”) for, among other things, non-disclosure of financial liabilities in the original KBL June 30, 2020 and September 30, 2020 Quarterly Reports on Form 10-Q of the matters disclosed in Note 14 (as restated) of the Company’s September 30, 2020 financial statements in the Company’s amended Quarterly Report on Form 10-Q filed on February 5, 2021. The Company is seeking damages resulting from discrepancies in the financial statements of KBL, unauthorized monetary transfers by Dr. Krauss, and inappropriate redemption of the shares associated with KBL. There can be no assurance that the Company will be successful in its legal actions. As of December 31, 2021, the Company recorded a legal accrual of $250,000 to cover the legal expenses of the former executives of KBL.
F-34
On October 5, 2021, Dr. Krauss and the KBL Affiliates filed an Answer, Counterclaims and Third-Party Complaint (the “Krauss Counterclaims”) against the Company and twelve individuals who are, or were, directors and/or officers of the Company, i.e., Marc Feldmann, Lawrence Steinman, James N. Woody, Teresa DeLuca, Frank Knuettel II, Pamela Marrone, Lawrence Gold, Donald A. McGovern, Jr., Russell T. Ray, Richard W. Barker, Shoshana Shendelman and Ozan Pamir (collectively, the “Third-Party Defendants”). On October 27, 2021, the Company and Ozan Pamir filed an Answer to the Krauss Counterclaims, and all of the other Third-Party Defendants filed a Motion to Dismiss as to the Third-Party Complaint.
On January 28, 2022, in lieu of filing an opposition to the Motion to Dismiss, Dr. Krauss and her affiliate companies filed a Motion for leave to file amended counterclaims and third-party complaint, and to dismiss six of the current and former directors previously named, i.e., to dismiss Teresa DeLuca, Frank Knuettel II, Pamela Marrone, Russell T. Ray, Richard W. Barker and Shoshana Shendelman. The Motion was granted by stipulation and, on February 24, 2022, Dr. Krauss filed an amended Answer, Counterclaims and Third-Party Complaint (the “Amended Counterclaims”). In essence, the Amended Counterclaims allege (a) that the Company and the remaining Third-Party Defendants breached fiduciary duties to Dr. Krauss by making alleged misstatements against Dr. Krauss in SEC filings and failing to register her shares in the Company so that they could be traded, and (b) the Company breached contracts between the Company and Dr. Krauss for registration of such shares, and also failed to pay to Dr. Krauss the amounts alleged to be owing under a promissory note in the principal amount of $371,178, plus an additional $300,000 under Dr. Krauss’ resignation agreement. The Amended Counterclaims seek unspecified amounts of monetary damages, declaratory relief, equitable and injunctive relief, and attorney’s fees and costs.
On March 16, 2022, Donald A. McGovern, Jr. and Lawrence Gold filed a Motion to Dismiss the Amended Counterclaims against them, and the Company and the remaining Third-Party Defendants filed an Answer to the Amended Counterclaims denying the same. The Company and the Third-Party Defendants intend to continue to vigorously defend against all of the Amended Counterclaims, however, there can be no assurance that they will be successful in the legal defense of such Amended Counterclaims.
Action Against the Company by Dr. Krauss
On August 19, 2021, Dr. Krauss initiated legal action in the Chancery Court of Delaware against the Company. The original Complaint sought expedited relief and made the following two claims: (1) it alleged that the Company is obligated to advance expenses including, attorney's fees, to Dr. Krauss for the costs of defending against the SEC and certain Subpoenas served by the SEC on Dr. Krauss; and (2) it alleged that the Company is also required to reimburse Dr. Krauss for the costs of bringing this lawsuit against the Company. On or about September 3, 2021, Dr. Krauss filed an Amended and Supplemental Complaint (the “Amended Complaint”) in this action, which added the further claims that Dr. Krauss is also allegedly entitled to advancement by the Company of her expenses, including attorney’s fees, for the costs of defending against the Third-Party Complaint in the Tyche action referenced below, and the costs of defending against the Company’s own Complaint against Dr. Krauss as described above. On or about September 23, 2021, the Company filed its Answer to the Amended Complaint in which the Company denied each of Dr. Krauss’ claims and further raised numerous affirmative defenses with respect thereto.
On November 15, 2021, Dr. Krauss filed a Motion for Summary Adjudication as to certain of the issues in the case, which was opposed by the Company. A hearing on such Motion was held on December 7, 2021, and, on March 7, 2022, the Court issued a decision in the matter denying the Motion for Summary Adjudication in part and granting it in part. The Court then issued an Order implementing such a decision on March 29, 2022. The parties will now engage in proceedings set forth in that implementing Order. No monetary amounts have yet been determined as to any sums that the Company may be required to pay to Dr. Krauss in this regard.
F-35
Action Against Tyche Capital LLC
This is a litigation case which the Company commenced and filed against defendant Tyche Capital LLC (“Tyche”) in the Supreme Court of New York, in the County of New York, on April 15, 2021. In its Complaint, the Company alleged claims against Tyche arising out of Tyche’s breach of its written contractual obligations to the Company as set forth in a “Guarantee And Commitment Agreement” dated July 25, 2019, and a “Term Sheet For KBL Business Combination With CannBioRex” dated April 10, 2019 (collectively, the “Subject Guarantee”). The Company alleges in its Complaint that, notwithstanding demand having been made on Tyche to perform its obligations under the Subject Guarantee, Tyche has failed and refused to do so, and is currently in debt to the Company for such failure in the $6,776,686, together with interest accruing thereon at the rate set forth in the Subject Guarantee.
On or about May 17, 2021, Tyche responded to the Company's Complaint by filing an Answer and Counterclaims against the Company alleging that it was the Company, rather than Tyche, that had breached the Subject Guarantee. Tyche also filed a Third-Party Complaint against six third-party defendants, including three members of the Company's management, Sir Marc Feldman, Dr. James Woody, and Ozan Pamir (collectively, the “Individual Company Defendants”), claiming that they allegedly breached fiduciary duties to Tyche with regards to the Subject Guarantee. In that regard, on June 25, 2021, each of the Individual Company Defendants filed a Motion to Dismiss Tyche’s Third-Party Complaint against them.
On November 23, 2021, the Court granted the Company’s request to issue an Order of attachment against all of Tyche’s shares of the Company’s stock that had been held in escrow. In so doing, the Court found that the Company had demonstrated a likelihood of success on the merits of the case based on the fact alleged in the Company’s Complaint.
On February 18, 2022, Tyche filed an Amended Answer, Counterclaims and Third-Party Complaint. On March 22, 2022, the Company and each of the Individual Company Defendants filed a Motion to Dismiss all of Tyche's claims. The Company and the Individual Company Defendants intend to continue to vigorously defend against all of Tyche's claims, however, there can be no assurance that they will be successful in the legal defense of such claims.
Cantor Fitzgerald & Co. Breach of Contract
Cantor Fitzgerald & Co. (“Cantor”) initiated legal action against the Company in the Supreme Court of New York, County of New York on April 22, 2021, alleging causes of action against the Company arising out of a written Fee Agreement dated February 27, 2018. The Complaint alleged that, pursuant to the Fee Agreement and services performed by Cantor thereunder, the Company became indebted to Cantor in the amount of $1,500,000. The Complaint further alleged that Cantor and the Company subsequently entered into a written Settlement And Release Agreement whereby Cantor agreed to release its monetary claims against the Company in exchange for issuance to Cantor of 150,000 shares of common stock of the Company. The Complaint acknowledged Cantor’s receipt of such common stock from the Company, but alleged that the Company failed to timely file a Registration Statement with the Securities and Exchange Commission, thereby damaging Cantor’s rights relative to the issued shares of common stock. The Complaint asked to declare the Settlement And Release Agreement null and void, and to cause the Company to pay to Cantor the sum of $1,500,000. The Company never appeared in the case nor responded to the Complaint because the Company and Cantor entered into a Mutual Settlement and General Release Agreement effective as of October 12, 2021 (the “Cantor Settlement”). The Company performed all of its obligations under the Cantor Settlement by paying the sum of $200,000, and the Cantor Settlement resolved and released all issues by and between the Company and Cantor. As a result thereof, Cantor dismissed the entirety of the case with prejudice on October 21, 2021.
F-36
Action Against Ronald Bauer & Samantha Bauer
The Company and two of its wholly-owned subsidiaries, Katexco Pharmaceuticals Corp. and CannBioRex Pharmaceuticals Corp. (collectively, the “Company Plaintiffs”), initiated legal action against Ronald Bauer and Samantha Bauer, as well as two of their companies, Theseus Capital Ltd. and Astatine Capital Ltd. (collectively, the “Bauer Defendants”), in the Supreme Court of British Columbia on February 25, 2022, 2022. The Company Plaintiffs are seeking damages against the Bauer Defendants for misappropriated funds and stock shares, unauthorized stock sales, and improper travel expenses, in the combined sum of at least $4,395,000 CAD [$3,460,584 USD] plus the additional sum of $2,721,036 USD. The Company Plaintiffs are currently seeking to effect service of process on each of the Bauer Defendants. Service of process has been effected on the Bauer Defendants, however, their responses are not yet due. There can be no assurance that the Company Plaintiffs will be successful in this legal action.
EarlyBird Settlement Agreement
On October 17, 2018, KBL entered into an agreement with EarlyBird-Capital, Inc. (“EarlyBird”), whereby EarlyBird would introduce potential targets to the Company on a non-exclusive basis for the purpose of consummating a merger, capital stock exchange, asset acquisition, or other similar business combination. Upon the closing of a transaction, the Company will pay EarlyBird a finder’s fee, payable in cash, of 1% of the value of the transaction, minus any liabilities at closing in excess of $5,000,000. On April 23, 2021, the Company settled the amounts due pursuant to a certain finder agreement entered into with EarlyBird on October 17, 2017 (the “Finder Agreement”). The Company’s Board of Directors determined it was in the best interests to settle all claims which had been made or could be made with respect to the Finder Agreement and entered into a settlement agreement (the “Settlement Agreement”). Pursuant to the Settlement Agreement, the Company paid EarlyBird a cash payment of $275,000 and issued 225,000 shares of the Company’s restricted common stock to EarlyBird valued at $1,973,250.
Yissum Research and License Agreement
On May 13, 2018, CBR Pharma entered into a worldwide research and license agreement with Yissum Research Development Company of the Hebrew University of Jerusalem, Ltd. (“Yissum Agreement”) allowing CBR Pharma to utilize certain patent (the “Licensed Patents”). The Licensed Patents shall expire, if not earlier terminated pursuant to the provisions of the Yissum Agreement, on a country-by-country, product-by-product basis, upon the later of: (i) the date of expiration in such country of the last to expire Licensed Patent included in the Licensed Technology; (ii) the date of expiration of any exclusivity on the product granted by a regulatory or government body in such country; or (iii) the end of a period of twenty (20) years from the date of the First Commercial Sale in such country. Should the periods referred to in items (i) or (ii) above expire in a particular country prior to the period referred to in item (iii), above, the license in that country or those countries shall be deemed a license to the Know-How during such post-expiration period.
Royalties will be payable to Yissum if sales of any products which use, exploit or incorporate technology covered by the Licensed Patents (“Net Sales”) are US $500,000,000 or greater, calculated at 3% for the first annual $500,000,000 of Net Sales and at 5% of Net Sales thereafter.
F-37
Pursuant to the Yissum Agreement, if Yissum achieves the following milestones, CBR Pharma will be obligated to make the following payments:
i) $75,000 for successful point of care in animals;
ii) $75,000 for submission of the first investigational new drug testing;
iii) $100,000 for commencement of one phase I/II trial;
iv) $150,000 for commencement of one phase III trial;
v) $100,000 for each product market authorization/clearance (maximum of $500,000); and
vi) $250,000 for every $250,000,000 in accumulated sales of the product until $1,000,000,000 in sales is achieved.
In the event of an exit event (“Event”), which may be defined as either, a transaction or series of transactions under which the receipt of any consideration, monetary or otherwise by the Company or its shareholders is received in consideration for the sale of shares of the Company or shareholders, or an initial public offering (“IPO”) of the Company, but for greater certainty excludes a reorganization of the Company where the ultimate equity holders of the reorganized entity remain substantially the same as that of the Company, the Company will issue 5% of the issued and outstanding shares, on a fully diluted basis, to Yissum prior to the closing of an Event. These shares will be subject to: (a) as to half of such shares, a lock-up period ending 12 months from the Event date and as to the other half of such shares, a lock-up period ending 24 months from the Event date, and (b) in any event, any resale restrictions (including lock-ups and hold periods). See Note 13 – Stockholders’ Equity (Deficiency) for more information on the shares issued to Yissum as part of the business combination.
CBR Pharma is also party to consulting agreements with Yissum, whereby Yissum has agreed to provide two of its employees as consultants to the Company for $100,000 per annum per person for a term of three years, commencing May 13, 2018.
On January 1, 2020, CBR Pharma entered into a first amendment to the Yissum Agreement (“First Amendment”) with Yissum, allowing CBR Pharma to sponsor additional research performed by two Yissum professors. Pursuant to the terms of the First Amendment, the Company will pay Yissum $200,000 per year plus 35% additional for University overhead for the additional research performed by each professor over an 18-month period, starting May 1, 2019. As of December 31, 2021, the Company owes no outstanding balance in connection with the Yissum Agreement (as amended). As of December 31, 2020, the Company owed an outstanding balance of $418,098 in connection with the Yissum Agreement (as amended), of which $48,908 was reflected within accounts payable and $370,000 was included in accrued expenses on the accompanying consolidated balance sheet. During the years ended December 31, 2021 and 2020, the Company recognized research and development expenses of $443,151 and $442,453, respectively, related to this agreement.
Additional Yissum Agreement
On November 11, 2019 (the “Effective Date”), CBR Pharma entered into a new worldwide research and license agreement with Yissum (the “Additional Yissum Agreement”), allowing CBR Pharma to obtain a license and perform the research, development and commercialization of the licensed patents (the “Licensed Patents”) in the research of cannabinoid salts relating to arthritis and pain management. Within 60 days after the end of the first anniversary of the Effective Date, Yissum will present the Company with a detailed written report summarizing the results of their research.
The Licensed Patents shall expire, if not earlier terminated pursuant to the provisions of the Additional Yissum Agreement, on a country-by-country, product-by- product basis, upon the later of: (i) the date of expiration in such country of the last to expire Licensed Patent included in the Licensed Technology; (ii) the date of expiration of any exclusivity on the product granted by a regulatory or government body in such country; or (iii) the end of a period of twenty (20) years from the date of the first commercial sale in such country. Should the periods referred to in items (i) or (ii) above expire in a particular country prior to the period referred to in item (iii), above, the license in that country or those countries shall be deemed a license to the know-how during such post-expiration period.
Pursuant to the terms of the Additional Yissum Agreement, CBR Pharma paid Yissum a non-refundable license fee of $70,000 and will pay an aggregate of $398,250 of research, development and consulting fees over the term of the Additional Yissum Agreement, as well as an annual license maintenance fee of $25,000, beginning on the first anniversary of the Effective Date.
F-38
The Company shall pay Yissum the following amounts in connection with the achievement of the following milestones:
| ● | Submission of the first Investigational New Drug application: $75,000 |
| ● | Dosing of first patient in phase II trial: $100,000 |
| ● | Dosing of first patient in phase Ill trial: $150,000 |
| ● | Upon first market authorization/clearance: $150,000 |
| ● | Upon second market authorization/clearance: $75,000 |
| ● | For every $250,000,000.00 US in accumulated Net Sales of the Product until $1,000,000,000.00 US in sales: $250,000 |
Upon the commercialization of the license, the Company shall pay Yissum a royalty equal to 3% of the first aggregate $500,000,000 of annual net sales and 5% thereafter. As of December 31, 2021, the Company had no balances in accounts payable and accrued expenses, respectively, relating to the Additional Yissum Agreement; as of December 31, 2020, the Company had $91,748 and $298,686 of accounts payable and accrued expenses, respectively, relating to the Additional Yissum Agreement. During the years ended December 31, 2021 and 2020, the Company recorded the purchase of the patents of $0 and $72,995, respectively, as an intangible asset to be amortized on a straight-line basis over the remaining lives of the patents and $246,753 and $477,411, respectively, of research and development expenses.
Evotec Agreement
On June 7, 2018, Katexco entered into an agreement (the “Drug Discovery Services Agreement”) with Evotec International GmbH (“Evotec”), whereby the Company and Evotec have agreed to negotiate research programs to be conducted by Evotec for the Company. Pursuant to the Drug Discovery Services Agreement, Evotec has agreed to conduct specified research services (the “Project”). The Project is scheduled to be conducted over a 24-month period, over which the Company will fund a minimum of $4,937,500 and a maximum of $5,350,250, based on quarterly invoices. During the years ended December 31, 2021 and 2020, the Company expensed $0 and $31,979, respectively, of research and development expenses in connection with the Drug Discovery Services agreement, and recorded interest expense of $0 and $31,979, respectively, on unpaid balances owed related to the Drug Discovery Services Agreement, which is included in accounts payable on the accompanying consolidated balance sheets. As of December 31, 2021 and 2020, unpaid balances owed related to the Drug Discovery Services Agreement amounted to $0 and $1,342,299, respectively.
Stanford License Agreement
On May 8, 2018, Katexco entered into a six-month option agreement (the “Stanford Option”) with Stanford University (“Stanford”) under which Stanford granted the Company a six-month option to acquire an exclusive license for patents (the “Licensed Patents”) which are related to biological substances used to treat auto- immune diseases. In consideration for the Stanford Option, the Company paid Stanford $10,000 (the “Option Payment”), which was creditable against the first anniversary license maintenance fee payment.
On July 25, 2018, Katexco exercised their six-month option and entered into an exclusive license agreement with equity (the “Stanford License Agreement”) with Stanford. Pursuant to the Stanford License Agreement, beginning upon the first anniversary of the effective date, and each anniversary thereafter, the Company will pay Stanford, in advance, a yearly license maintenance fee of $20,000, on each of the first and second anniversaries and $40,000 on each subsequent anniversary, which will be expensed on a straight-line basis annually.
F-39
Furthermore, the Company will be obligated to make the following milestone payments:
i) $100,000 upon initiation of Phase II trial,
ii) $500,000 upon the first U.S. Food and Drug Administration approval of a product (the “Licensed Product”) resulting from the Licensed Patents; and
iii) $250,000 upon each new Licensed Product thereafter.
The Stanford License Agreement is cancellable by the Company with 30 days’ notice. Royalties, calculated at 2.5% of 95% of net product sales, will be payable to Stanford. Also, the Company will reimburse Stanford for patent expenses as per the agreement. The Company paid Stanford $20,000 for the annual license maintenance fee that was recorded to prepaid expenses and is being expensed on a straight-line basis over 12 months, which had a zero balance as of December 31, 2021. During the years ended December 31, 2021 and 2020, the Company recorded patent and license fees of $78,245 and $32,979, respectively, related to the Stanford License Agreement, which is included in general and administrative expenses on the accompanying statements of operations and comprehensive loss.
Oxford University Agreements
On March 22, 2019, 180 LP entered into a one-year research agreement (the “Research Agreement”) with Oxford pursuant to which 180 LP agreed to pay Oxford approximately $900,000 to perform certain research and to obtain the exclusive option to negotiate a license to commercially exploit any arising intellectual property as a result of Oxford’s research. During the year ended December 31, 2020, the Company recognized research and development expenses of $186,391. During the year ended December 31, 2019, the Company recognized research and development expenses of $396,950 related to the Research Agreement. Because 180 LP is an accounting acquiree in the Reorganization, the contract expense included in the accompanying consolidated statements of operations and comprehensive loss is only for the post-Reorganization period.
On September 18, 2020, CBR Pharma entered into a 3 year research and development agreement (the “3 Year Oxford Agreement”) with Oxford to research and investigate the mechanisms underlying fibrosis in exchange for aggregate consideration of $1,085,738 (£795,468), of which $109,192 (£80,000) is to be paid 30 days after the project start date and the remaining amount is to be paid in four equal installments of $244,136 (£178,867) on the six month anniversary and each of the annual anniversaries of the project start date. The agreement can be terminated by either party upon written notice or if the Company remains in default on any payments due under this agreement for more than 30 days. During the year ended December 31, 2021 and 2020, the Company recognized $364,673 (£264,938) and $113,433 (£88,385), respectively, of research and development expenses in connection with the 3 Year Oxford Agreement.
On September 21, 2020, CBR Pharma entered into a 2 year research and development agreement (the “2 Year Oxford Agreement”) with Oxford University for the clinical development of cannabinoid drugs for the treatment of inflammatory diseases in exchange for aggregate consideration of $625,124 (£458,000), of which $138,917 (£101,778) is to be paid 30 days after the project start date and the remaining amount is to be paid every 6 months after the project start date in 4 installments, whereby $138,917 (£101,778) is to be paid in the first 3 installments and $69,456 (£50,888) is to be paid as the final installment. The agreement can be terminated by either party upon written notice or if the Company remains in default on any payments due under this agreement for more than 30 days. During the years ended December 31, 2021 and 2020, the Company recognized $139,977 (£101,778) and $78,374 (£61,067) of research and development expenses, respectively, in connection with the 2 Year Oxford Agreement, which is reflected within accrued expenses on the accompanying consolidated balance sheet.
As of December 31, 2021, the Company owed Oxford an aggregate of $0 for the 2-year agreement. As of December 31, 2020, the Company owed Oxford an aggregate of $704,960, including $693,515 of accounts payable and $11,445 of accrued expense.
F-40
On May 24, 2021, the Company entered into a research agreement with the University of Oxford (“Oxford” and the “Fifth Oxford Agreement”), pursuant to which the Company will sponsor work at the University of Oxford to conduct a multi-center, randomized, double blind, parallel group, feasibility study of anti-TNF injection for the treatment of adults with frozen shoulder during the pain-predominant phase. As consideration, the Company agreed to make the following payments to Oxford:
| Amount Due | ||||
| Milestone | (excluding VAT) | |||
| Upon signing of the Fifth Oxford Agreement | £ | 70,546 | ||
| 6 months post signing of the Fifth Oxford Agreement | £ | 70,546 | ||
| 12 months post signing of the Fifth Oxford Agreement | £ | 70,546 | ||
| 24 months post signing of the Fifth Oxford Agreement | £ | 70,546 | ||
The Company paid the first milestone of $97,900 (£70,546) on September 3, 2021, which was due upon signing of the Fifth Oxford Agreement, which was recorded to prepaid expenses and will be amortized over the term of the agreement on a straight-line basis. During the year ended December 31, 2021, the Company recorded $210,215 (£152,848) of research and development expenses and has a prepaid balance of $80,852 (£58,788) related to the Fifth Oxford Agreement.
On November 2, 2021, the Company and Oxford University entered into a twenty-year licensed technology agreement of the HMGB1 molecule, which is related to tissue regeneration, whereby Oxford University agreed to license the technology to the Company for research, development and use of the licensed patents. The Company agreed to pay Oxford University for past patent costs $66,223 (£49,207), an initial License fee of $13,458 (£10,000), future royalties based on sales and milestones, and an annual maintenance fee of $4,037 (£3,000). The Company has the option to terminate the agreement after the third anniversary of the agreement.
Kennedy License Agreement
On September 27, 2019, 180 LP entered into a license agreement (the “Kennedy License Agreement”) with the Kennedy Trust for Rheumatology Research (“Kennedy”) exclusively in the U.S., Japan, United Kingdom and countries of the EU, for certain licensed patents (the “Kennedy Licensed Patents”), including the right to grant sublicenses, and the right to research, develop, sell or manufacture any pharmaceutical product (i) whose research, development, manufacture, use, importation or sale would infringe the Kennedy Licensed Patents absent the license granted under the Kennedy License Agreement or (ii) containing an antibody that is a fragment of or derived from an antibody whose research, development, manufacture, use, importation or sale would infringe the Kennedy Licensed Patents absent the license granted under the Kennedy License Agreement, for all human uses, including the diagnosis, prophylaxis and treatment of diseases and conditions.
F-41
As consideration for the grant of the Kennedy Licensed Patents, 180 LP paid Kennedy an upfront fee of GBP £60,000, (USD $74,000) on November 22, 2019, which was recognized as an intangible asset for the purchase of the licensed patents and is being amortized over the remaining life of the patents. 180 LP will also pay Kennedy royalties equal to (i) 1% of the net sales for the first annual GBP £1 million (USD $1,283,400) of net sales, and (ii) 2% of the net sales after the net sales are at or in excess of GBP £1 million, as well as 25% of all sublicense revenue, provided that the amount of such percentage of sublicense revenue based on amounts which constitute royalties shall not be less than 1% on the first cumulative GBP £1 million of net sales of the products sold by such sublicenses or their affiliates, and 2% on that portion of the cumulative net sales of the products sold by such sublicenses or their affiliates in excess of GBP £1 million.
The term of the royalties paid by the Company to Kennedy will expire on the later of (i) the last valid claim of a patent included in the Kennedy Licensed Patents which covers or claims the exploitation of a product in the applicable country; (ii) the expiration of regulatory exclusivity for the product in the country; or (iii) 10 years from the first commercial sale of the product in the country. The Kennedy License Agreement may be terminated without cause by providing a 90-day notice.
Petcanna Sub-License Agreement
On August 20, 2018, CBR Pharma entered into a sub-license agreement (the “Sub-License Agreement”) with its wholly owned subsidiary, Petcanna Pharma Corp. (“Petcanna”), of which the Company’s former Chief Financial Officer is a director. Petcanna is a private company with one common principal with the Company.
Pursuant to the terms of the Sub-license Agreement, the Company has granted a sub-license on the Licensed Patents to pursue development and commercialization for the treatment of any and all veterinary conditions. In consideration, Petcanna will (a) issue 9,000,000 common shares of its share capital (the “Petcanna Shares”) 30 days after the effective date; and (b) pay royalties of 1% of net sales. The Company will be issued 85% and Yissum will be issued 15% of the 9,000,000 common shares of the Petcanna subsidiary. The Petcanna shares are deemed to be founders shares with no value. The Petcanna shares have not been issued as of December 31, 2021, due to administrative delays.
360 Life Sciences Corp. Agreement - Related Party (Acquisition of ReFormation Pharmaceuticals Corp.)
On July 1, 2020, the Company entered into an amended agreement with ReFormation Pharmaceuticals, Corp. (“ReFormation”) and 360 Life Sciences Corp. (“360”), whereby 360 has entered into an agreement to acquire 100% ownership of ReFormation, on or before July 31, 2020 (“Closing Date”). The Company shares officers and directors with each of ReFormation and 360. Upon the Closing Date, 360 will make tranche payments in tranches to 180 LP in the aggregate amount of $300,000. The parties agree that the obligations will be paid by 360 to 180 LP by payments of $100,000 for every $1,000,000 raised through the financing activities of 360, up to a total of $300,000, however, not less than 10% of all net financing proceeds received by 360 shall be put towards the obligation to the Company until paid in full. This transaction closed on July 31, 2020.
On February 26, 2019, 180 LP entered into a one-year agreement (the “Pharmaceutical Agreement”) with ReFormation, a related party that shares directors and officers of 180 LP, pursuant to which the ReFormation agreed to pay 180 LP $1.2 million for rights of first negotiation to provide for an acquisition of any arising intellectual property or an exclusive licensing, partnering, or collaboration transaction to use any arising intellectual property with respect to a contemplated research agreement between the Company and Oxford (see Oxford University Agreements, above), which was signed on March 22, 2019 and therefore is the start date of the project. Of the $1.2 million receivable from Reformation pursuant to the Pharmaceutical Agreement, $0.9 million was received by the Company on March 14, 2019 and the remaining $0.3 million will be received over the one-year term of the agreement.
F-42
180 LP is recognizing the income earned in connection with the Pharmaceutical Agreement on a straight-line basis over the term of the agreement. During the years ended December 31, 2021 and 2020, 180 LP recognized $0 and $240,000, respectively, of income related to the Pharmaceutical Agreement, which is included in other income in the accompanying consolidated statement of operations and other comprehensive income loss. As of December 31, 2021, the Company charged the $300,000 receivable to bad debt expense. As of December 31, 2020, the Company had a receivable of $300,000 representing income earned in excess of cash received pursuant to the Pharmaceutical Agreement.
Operating Leases
On February 17, 2020, the Company entered into a twelve-month lease agreement to lease office space located in London, UK. The rent is approximately GBP £4,250 (USD $5,801) per month over the lease term for a total lease commitment of GBP £61,200 (USD $83,532). The lease commenced on February 19, 2020 and expires on February 18, 2021. In connection with the lease, the Company paid the landlord a security deposit of GBP £5,100 (USD $6,961). The lease shall continue until one of the following two events occur: (a) another lease agreement is entered into by the parties or (b) either party gives not less than three full calendar months written notice terminating this agreement prior to the expiration date. The Company terminated the lease in August 2020 due to the COVID-19 pandemic. The Company’s rent expense amounted to $0 and GBP £30,257 (USD $38,831) for the years ended December 31, 2021 and 2020, respectively. Rent expense is reflected in general and administrative expenses in the consolidated statements of operations and comprehensive loss.
Consulting Agreement
Related Party Consulting Agreement
On February 22, 2021, the Company entered into a consultancy agreement (as amended, the “Consulting Agreement”) with a related party, Prof. Jagdeep Nanchahal (the “Consultant”). The Consulting Agreement was effective December 1, 2020.
Pursuant to the Consulting Agreement, the Company agreed to pay the Consultant 15,000 British Pounds (GBP) per month (approximately $20,800) during the term of the agreement, increasing to 23,000 GBP per month (approximately $32,000) on the date (a) of publication of the data from the phase 2b clinical trial for Dupuytren’s Contracture (RIDD) and (b) the date that the Company has successfully raised over $15 million in capital. The Company also agreed to pay the Consultant the following bonus amounts:
| ● | the sum of £100,000 (approximately $138,000) upon submission of the Dupuytren’s Contracture clinical trial data for publication in a peer-reviewed journal (“Bonus 1”); |
| ● | the sum of £434,673 GBP (approximately $605,000) (“Bonus 2”), which is earned and payable upon the Company raising a minimum of $15 million in additional funding, through the sale of debt or equity, after December 1, 2020 (the “Vesting Date”). Bonus 2 is payable within 30 days of the Vesting Date and shall not be accrued, due or payable prior to the Vesting Date. Bonus 2 is payable, at the election of the Consultant, at least 50% (fifty percent) in shares of the Company’s common stock, at the lower of (i) $3.00 per share, or (ii) the trading price on the date of the grant, with the remainder paid in GBP; |
| ● | the sum of £5,000 (approximately $7,000) on enrollment of the first patient to the phase 2 frozen shoulder trial (“Bonus 3”); and |
| ● | the sum of £5,000 (approximately $7,000) for enrollment of the first patient to the phase 2 delirium/POCD trial (“Bonus 4”). |
The Consulting Agreement has an initial term of three years, and renews thereafter for additional three-year terms, until terminated as provided in the agreement. The Consulting Agreement can be terminated by either party with 12 months prior written notice (provided the Company’s right to terminate the agreement may only be exercised if the Consultant fails to perform his required duties under the Consulting Agreement), or by the Company immediately under certain conditions specified in the Consulting Agreement if (a) the Consultant fails or neglects efficiently and diligently to perform the services required thereunder or is guilty of any breach of its or his obligations under the agreement (including any consent granted under it); (b) the Consultant is guilty of any fraud or dishonesty or acts in a manner (whether in the performance of the services or otherwise) which, in the reasonable opinion of the Company, has brought or is likely to bring the Consultant, the Company or any of its affiliates into disrepute or is convicted of an arrestable offence (other than a road traffic offence for which a non-custodial penalty is imposed); or (c) the Consultant becomes bankrupt or makes any arrangement or composition with his creditors. If the Consulting Agreement is terminated by the Company for any reason other than cause, the Consultant is entitled to a lump sum payment of 12 months of his fee as of the date of termination.
F-43
Effective March 30, 2021, in satisfaction of amounts owed to the Consultant for 50% of Bonus 2, the Company issued 100,699 shares of the Company’s common stock to the Consultant. Additionally, on April 15, 2021, in satisfaction of amounts owed to the Consultant for an additional 19% of Bonus 2, the Company issued 37,715 of the Company’s common stock to the Consultant.
Effective August 27, 2021, in satisfaction of amounts owed to the Consultant for the remainder of Bonus 2, the Company issued 61,535 shares of the Company’s common stock to the Consultant since the Company raised $15 million in a financing transaction, as per the agreement. All issuances were made under the Company’s 2020 Omnibus Incentive Plan. See Note 13 – Stockholders’ Equity.
In December 2021, the Dupuytren’s Contracture clinical trial data was submitted for publication in a peer-reviewed journal and Bonus 1 was paid to the Consultant.
Larsen Consulting Agreement
On April 29, 2021, the Company entered into a consulting agreement with Glenn Larsen, the former Chief Executive Officer of 180 LP, to act in the capacity as negotiator for the licensing of four patents. In consideration for services provided, the Company agreed to compensate Mr. Larsen with $50,000 of its restricted common stock (valued based on the closing sales price of the Company’s common stock on the date the Board of Directors approved the agreement, which shares have not been issued to date). The fully vested shares will be issued to Mr. Larsen pursuant to the 2020 Omnibus Incentive Plan, upon the Company entering into a licensing transaction with the assistance of Mr. Larsen. On November 2, 2021, the Company and Oxford University entered into a license agreement and therefore 5,423 shares were issued to Mr. Larsen on November 3, 2021 pursuant to the Company’s 2020 Omnibus Incentive Plan.
Employment Agreement of Chief Executive Officer
On February 25, 2021, the Company entered into an amended agreement with Dr. James Woody, the Chief Executive Officer of the Company (the “CEO”) (the “A&R Agreement”), dated February 24, 2021, and effective November 6, 2020, which replaced the CEO’s prior agreement with the Company. Pursuant to the A&R Agreement, the CEO agreed to serve as an officer of the Company for a term of three years, which is automatically renewable thereafter for additional one-year periods, unless either party provides the other at least 90 days written notice of their intent to not renew the agreement. The CEO’s annual base salary under the agreement will initially be $450,000 per year, with automatic increases of 5% per annum.
As additional consideration for the CEO agreeing to enter into the agreement, the Company awarded him options to purchase 1,400,000 shares of the Company’s common stock, which have a term of 10 years, and an exercise price of $4.43 per share (the closing sales price on the date the board of directors approved the grant (February 26, 2021)). The options as subject to the Company’s 2020 Omnibus Incentive Plan and vest at the rate of (a) 1/5th of such options on the grant date; and (b) 4/5th of such options vesting ratably on a monthly basis over the following 36 months on the last day of each calendar month; provided, however, that such options vest immediately upon the CEO’s death or disability, termination without cause or a termination by the CEO for good reason (as defined in the agreement), a change in control of the Company or upon a sale of the Company.
F-44
The CEO is also eligible to receive an annual bonus, with a target bonus equal to 45% of his then-current base salary, based upon the Company’s achievement of performance and management objectives as set and approved by the Board of Directors and/or Compensation Committee in consultation with the CEO. At the CEO’s option, the annual bonus can be paid in cash or the equivalent value of the Company’s common stock or a combination. The Board of Directors, as recommended by the Compensation Committee, may also award the CEO bonuses from time to time (in stock, options, cash, or other forms of consideration) in its discretion. Under the A&R Agreement, the CEO is also eligible to participate in any stock option plans and receive other equity awards, as determined by the Board of Directors from time to time. As of December 31, 2021, the Company recorded $205,500 of accrued bonus payable to the CEO.
The A&R agreement can be terminated any time by the Company for cause (subject to the cure provisions of the agreement), or without cause (with 60 days prior written notice to the CEO), by the CEO for good reason (as described in the agreement, and subject to the cure provisions of the agreement), or by the CEO without good reason. The agreement also expires automatically at the end of the initial term or any renewal term if either party provides notice of non-renewal as discussed above.
In the event the A&R Agreement is terminated without cause by the Company, or by the CEO for good reason, the Company agreed to pay him the lesser of 18 months of salary or the remaining term of the agreement, the payment of any accrued bonus from the prior year, his pro rata portion of any current year’s bonus and health insurance premiums for the same period that he is to receive severance payments (as discussed above).
The A&R Agreement contains standard and customary invention assignment, indemnification, confidentiality and non-solicitation provisions, which remain in effect for a period of 24 months following the termination of his agreement.
Employment Agreement of Chief Financial Officer
On February 25, 2021, the Company entered into an Employment Agreement (the “CFO Agreement”) dated February 24, 2021, and effective November 6, 2020, with the Company’s Interim Chief Financial Officer, Ozan Pamir. Pursuant to the agreement, the CFO agreed to serve as the Interim Chief Financial Officer (“CFO”) of the Company for an initial salary of $300,000 per year, subject to increase to a mutually determined amount upon the closing of a new financing as well as annual increases.
As additional consideration for the CFO agreeing to enter into the agreement, the Company awarded him options to purchase 180,000 shares of the Company’s common stock, which have a term of 10 years, and an exercise price of $4.43 per share (the closing sales price on the date the board of directors approved the grant (February 26, 2021)). The options are subject to the Company’s 2020 Omnibus Incentive Plan and vest at the rate of (a) 1/5th of such options upon the grant date; and (b) 4/5th of such options vesting ratably on a monthly basis over the following 36 months on the last day of each calendar month; provided, however, that such options vest immediately upon the CFO’s death or disability, termination without cause or a termination by the CFO for good reason (as defined in the agreement), a change in control of the Company or upon a sale of the Company.
Under the agreement, the CFO is eligible to receive an annual bonus, in a targeted amount of 30% of his then salary, based upon the Company’s achievement of performance and management objectives as set and approved by the CEO, in consultation with the CFO. The bonus amount is subject to adjustment. The Board of Directors, as recommended by the Compensation Committee of the Company (and/or the Compensation Committee), may also award the CFO bonuses from time to time (in stock, options, cash, or other forms of consideration) in its discretion. Under the CFO Agreement, the CFO is also eligible to participate in any stock option plans and receive other equity awards, as determined by the Board of Directors from time to time. As of December 30, 2021, the Company recorded $90,000 of accrued bonus payable to the CFO.
F-45
The agreement can be terminated any time by the Company with or without cause with 60 days prior written notice and may be terminated by the CFO at any time with 60 days prior written notice. The agreement may also be terminated by the Company with six days’ notice in the event the agreement is terminated for cause under certain circumstances. Upon the termination of the CFO’s agreement by the Company without cause or by the CFO for good reason, the Company agreed to pay him three months of severance pay.
The agreement contains standard and customary invention assignment, indemnification, confidentiality and non-solicitation provisions, which remain in effect for a period of 24 months following the termination of his agreement.
Employment Agreement of Chief Operating Officer/Chief Business Officer
On October 29, 2021, the Company entered into an Employment Agreement (the “COO/CBO Agreement”) dated October 27, 2021, and effective November 1, 2021, with Quan Vu. Pursuant to the agreement, Mr. Vu agreed to serve as the Chief Operating Officer/Chief Business Officer (“COO/CBO”) of the Company for an initial salary of $390,000 per year, subject to a $10,000 increase upon completion of a $50 Million financing and a yearly increase of five percent (5%) on each start-day anniversary.
As additional consideration for the COO/CBO agreeing to enter into the agreement, the Company awarded him options to purchase 275,000 shares of the Company’s common stock, which have a term of 10 years, and an exercise price equal to the Fair Market Value of the Company’s common stock on the date of grant which is still to be determined. The options are subject to the Company’s 2020 Omnibus Incentive Plan and vest ratably on a monthly basis over the following 48 months on the last day of each calendar month; provided, however, that such options vest immediately upon the COO/CBO death or disability, termination without cause or a termination by the COO/CBO for good reason (as defined in the agreement), a change in control of the Company or upon a sale of the Company.
Under the agreement, the COO/CBO is eligible to receive an annual bonus, in a targeted amount of 50% of his then salary, based upon the Company’s achievement of performance and management objectives as set and approved by the CEO, in consultation with the CFO. The annual bonus shall be paid on or before March 31 of the year following the year in which the bonus is earned. At the choice of the Executive, the annual bonus can be paid in cash or the equivalent value of the Company’s common stock or a combination of both. For calendar 2021, such Bonus payment, if any, will be prorated for approximately 2 months after the Start Date. The CEO, as approved by the Compensation Committee, may also award the Executive a bonus from time to time (in stock, options, cash, or other forms of consideration) in his discretion.
The agreement can be terminated any time by the Company with or without cause with 30 days prior written notice and may be terminated by the COO/CBO at any time with 30 days prior written notice. The agreement may also be terminated by the Company with ten days’ notice in the event the agreement is terminated for cause under certain circumstances. Upon the termination of the COO/CBO’s agreement by the Company without cause or by the COO/CBO for good reason, the Company agreed to pay him twelve months of severance pay, except if Executive separates from the Company prior to a one-year anniversary.
The agreement contains standard and customary invention assignment, indemnification, confidentiality and non-solicitation provisions, which remain in effect for a period of 24 months following the termination of his agreement.
NOTE 13 – STOCKHOLDERS’ EQUITY
Preferred Stock
Pursuant to the Company’s Second Amended and Restated Certificate of Incorporation filed on November 6, 2020, the Company has 5,000,000 preferred shares authorized at a par value of $0.0001 per share, of which 1,000,000 shares are designated as Series A Convertible Preferred Stock (“Series A Preferred”), 1 share is designated as the Class K Special Voting Share and 1 share is designated as the Class C Special Voting Share. The Class K Special Voting Share and the Class C Special Voting Share are together, the “Special Voting Shares”. As of December 31, 2020, there is no Series A Preferred issued or outstanding; there is one Class K Special Voting Share and one Class C special Voting Share issued and outstanding.
F-46
Series A Preferred Stock
The Series A Preferred is convertible into common stock at an initial conversion price of $5.28 per share, at the election of the holder, at any time following issuance, subject to certain anti-dilution adjustments. Upon a dilutive issuance (as defined) at a price per share lower than the existing conversion price, the conversion price will adjust to the lower of (a) the dilutive issuance price per share; or (b) the lowest volume-weighted-average-price during the five days preceding the dilutive issuance. Upon any conversion, a make-whole amount (as defined in the Certificate of Designation of the Series A Preferred) shall be due with respect to each share of Series A Preferred converted. At any time following the three-month anniversary of the Business Combination, the holder of the Series A Preferred had the right to force the Company to redeem all or any portion of the Series A Preferred then owned by the holder in cash. Series A Preferred stockholders were entitled to 10% dividends. Holders of the Series A Preferred had no voting rights.
The Company assumed 1,000,000 shares of issued and outstanding Series A Preferred in connection with the Business Combination with a carrying value of $1,411,265, which was net of a $1,922,068 discount from its stated value of $3,333,333. The discount included an original issuance discount of $333,333, cash issuance costs of $318,333, warrant issuance costs of $103,402 (fair value of warrant issued to placement agent), and a bifurcated redemption feature that was valued at $1,167,000 at issuance (see Note 9 – Derivative Liabilities). No accretion of the Series A Preferred discount was required because redemption wasn’t deemed to be probable.
On November 25, 2020, a dilutive issuance reduced the conversion price to the lower of (a) 96% of the lowest volume-weighted-average-price of the common stock during the five-day period preceding the conversion date; or (b) $5.28, both subject to a floor of $2.00 per share. The conversion price adjustment was treated as an extinguishment and reissuance of the outstanding Series A Preferred. On the extinguishment date, the bifurcated redemption feature was marked-to-market, increasing the value by $606,000 and recognizing a corresponding charge to change in fair value of derivative liabilities. The $1,411,265 carrying value of the preferred stock and the $1,773,000 fair value of the derivative liability were derecognized and we recognized the new $3,531,924 fair value of the preferred stock and the new $218,000 value of the bifurcated redemption feature. The difference of $565,659 was recognized as a deemed dividend.
During the period from November 30, 2020 to December 18, 2020, the 1,000,000 shares of Series A Preferred of the Company with a total conversion value of $3,666,667 were converted into shares of the Company’s common stock at conversion prices of between $2.00 and $2.31 per share, pursuant to the terms of such preferred stock. The bifurcated redemption features were marked-to-market just prior to each conversion, resulting in an aggregate charge of $42,068 to change in fair value of derivative liabilities and the $260,068 fair value of the bifurcated redemption features were derecognized on the conversion dates. At conversion, the aggregate $3,531,924 carrying value of the preferred stock and the $260,068 fair value of the derivative liability were derecognized and we recognized the $4,349,035 fair value of the 1,614,144 shares of common stock issued. The difference of $557,043 was recorded as deemed dividend expense, including $333,333 associated with the make-whole premiums and $223,710 associated with the contingent beneficial conversion feature. Due to such conversions, the Company currently has no shares of Series A Preferred issued or outstanding.
The aggregate deemed dividend presented on the income statement is comprised of the $333,333 make-whole dividend, the $223,710 beneficial conversion feature and a $565,659 extinguishment loss associated with the conversion price adjustment.
Common Stock
The Company is authorized to issue 100,000,000 shares of the Company’s common stock with a par value of $0.0001 per share. Holders of the Company’s shares of the Company’s common stock are entitled to one vote for each share.
F-47
During the year ended December 31, 2020, the Company issued 12,324,300 shares of its common stock, of which 12,292 shares were issued for cash consideration of $72,500, 240,540 shares with a grant date value of $1,057,989 were issued as compensation, 1,519,628 shares of common stock were issued to the KBL shareholders upon the consummation of the Reverse Merger, 1,619,144 shares of common stock were issued upon the conversion of $4,349,035 of Series A Preferred Stock, 1,521,157 shares were issued upon the exchange of common stock equivalents associated with the Special Voting Shares, 482,894 shares were issued upon conversion of 180 debt and 6,928,645 shares were issued as a result of the reverse recapitalization.
Sale of Common Stock and Warrants in the February 2021 Private Offering
On February 19, 2021, the Company entered into a Securities Purchase Agreement with certain purchasers (the “Purchasers”), pursuant to which the Company agreed to sell an aggregate of 2,564,000 shares of common stock (the “PIPE Shares”) and warrants to purchase up to an aggregate of 2,564,000 shares of common stock (the “PIPE Warrants”), at a combined purchase price of $4.55 per share and PIPE Warrant (the “Offering”). Aggregate gross proceeds from the offering were approximately $11.7 million. Net proceeds to the Company from the offering, after deducting the placement agent fees and estimated offering expenses payable by the Company, were approximately $10.7 million.
The PIPE Warrants have an exercise price equal to $5.00 per share, were immediately exercisable and are subject to customary anti-dilution adjustments for stock splits or dividends or other similar transactions. However, the exercise price of the PIPE Warrants will not be subject to adjustment as a result of subsequent equity issuances at effective prices lower than the then-current exercise price. The PIPE Warrants are exercisable for 5 years following the closing date. The PIPE Warrants are subject to a provision prohibiting the exercise of such PIPE Warrants to the extent that, after giving effect to such exercise, the holder of such PIPE Warrant (together with the holder’s affiliates, and any other persons acting as a group together with the holder or any of the holder’s affiliates), would beneficially own in excess of 4.99% of the Company’s outstanding common stock (which may be increased to 9.99% on a holder by holder basis, with 61 days prior written consent of the applicable holder). The PIPE Warrants were determined to be liability-classified (see Note 9, Derivative Liabilities). Of the $968,930 of placement agent fees and offering expenses, $364,812 was allocated to the PIPE Shares and $604,118 was allocated to the PIPE Warrant. Because the PIPE Warrants are liability classified, the $604,118 allocated to the warrants was immediately expensed.
In connection with the offering, the Company also entered into a Registration Rights Agreement, dated as of February 23, 2021, with the Purchasers (the “Registration Rights Agreement”). Pursuant to the Registration Rights Agreement, the Company agreed to file a registration statement with the SEC on or prior to April 24, 2021 to register the resale of the PIPE Shares and the shares of common stock issuable upon exercise of the PIPE Warrants (the “PIPE Warrant Shares”), and to cause such registration statement to be declared effective on or prior to June 23, 2021 (or, in the event of a “full review” by the SEC, August 22, 2021). The Company was in default of the terms of the Registration Rights Agreement as the registration statement to register the PIPE Shares and PIPE Warrant Shares was not filed by April 24, 2021; provided that such registration statement has been filed. As a result of this default, the Company was required to pay damages to the Purchasers in the aggregate amount of $174,993 each month, up to a maximum of $583,310. The Company incurred $524,979 of damages during the year ended December 31, 2021, which amount was paid, and as a result the Company is no longer in default.
Bridge Note Conversions
During the first quarter of 2021, certain noteholders elected to convert bridge notes with an aggregate principal balance of $365,750 and an aggregate accrued interest balance of $66,633 into an aggregate of 158,383 shares of the Company’s common stock at a conversion price of $2.73 per share, pursuant to the terms of such notes. (see Note 11 - Convertible Notes Payable).
F-48
Convertible Note Conversions
During the first quarter of 2021, certain noteholders elected to convert certain convertible notes payable with an aggregate principal balance of $1,234,333 and an aggregate accrued interest balance of $105,850 into an aggregate of 467,123 shares of the Company’s common stock at conversion prices ranging from $2.45-$3.29 per share, pursuant to the terms of such notes. (see Note 11 - Convertible Notes Payable).
EarlyBird Settlement
On April 23, 2021, the Company settled the amounts due pursuant to a certain finder agreement entered into with EarlyBird Capital, Inc. (“EarlyBird”) on October 17, 2017 (the “Finder Agreement”). The Company’s Board of Directors determined it was in the best interests to settle all claims which had been made or could be made with respect to the Finder Agreement and entered into a settlement agreement (the “Settlement Agreement”). Pursuant to the Settlement Agreement, the Company paid EarlyBird a cash payment of $275,000 and issued 225,000 shares of the Company’s restricted common stock with a grant date value of $1,973,250 to EarlyBird, in full satisfaction of accounts payable in the amount of $1,750,000. The Company recorded a loss of $223,250 in connection with the Settlement Agreement, which is included in (loss) gain on settlement of liabilities in the accompanying consolidated statements of operations.
Sale of Common Stock and Warrants in the August 2021 Offering
On August 23, 2021, the Company entered into a Securities Purchase Agreement with certain purchasers, pursuant to which the Company agreed to sell an aggregate of 2,500,000 shares of common stock and warrants to purchase up to an aggregate of 2,500,000 shares of common stock (the “August 2021 PIPE Warrants”), at a combined purchase price of $6.00 per share and August 2021 PIPE Warrant (the “August 2021 Offering”). Aggregate gross proceeds from the August 2021 Offering were approximately $15,000,000. Net proceeds to the Company from the August 2021 Offering, after deducting the placement agent fees and estimated offering expenses payable by the Company, were approximately $13.9 million.
The August 2021 PIPE Warrants have an exercise price equal to $7.50 per share, are immediately exercisable and are subject to customary anti-dilution adjustments for stock splits or dividends or other similar transactions. However, the exercise price of the August 2021 PIPE Warrants will not be subject to adjustment as a result of subsequent equity issuances at effective prices lower than the then-current exercise price. The August 2021 PIPE Warrants are exercisable for 5 years following the closing date. The August 2021 PIPE Warrants are subject to a provision prohibiting the exercise of such August 2021 PIPE Warrants to the extent that, after giving effect to such exercise, the holder of such August 2021 PIPE Warrant (together with the holder’s affiliates, and any other persons acting as a group together with the holder or any of the holder’s affiliates), would beneficially own in excess of 4.99% of the Company’s outstanding common stock (which may be increased to 9.99% on a holder by holder basis, with 61 days prior written consent of the applicable holder). Although the PIPE Warrants have a tender offer provision, the August 2021 PIPE Warrants were determined to be equity-classified because they met the limited exception in the case of a change-in-control. Because the August 2021 PIPE Warrants are equity-classified, the $1,120,000 of placement agent fees and offering expenses were fully accounted for as a reduction of additional paid in capital.
In connection with the August 2021 Offering, the Company also entered into a Registration Rights Agreement, dated as of August 23, 2021, with the purchasers (the “August 2021 Registration Rights Agreement”). Pursuant to the August 2021 Registration Rights Agreement, the Company agreed to file a registration statement with the SEC on or prior to September 12, 2021 to register the resale of the shares and the shares of common stock issuable upon exercise of the August 2021 PIPE Warrants (the “Warrant Shares”) sold in the August 2021 Offering, and to cause such registration statement to be declared effective on or prior to October 22, 2021 (or, in the event of a “full review” by the SEC, November 21, 2021). The registration statement was filed on August 31, 2021 and the SEC declared it effective on September 9, 2021, prior to the deadline set forth in the August 2021 Registration Rights Agreement.
F-49
Exchanges of Related Party Loans and Convertible Notes
On September 30, 2021, Dr. Lawrence Steinman and Sir Marc Feldmann, Ph.D., each of whom serve as Co-Executive Chairmen of the Company’s Board of Directors, agreed with the Company to convert amounts owed under outstanding loans with an aggregate principal balance of $693,371 and an aggregate accrued interest balance of $157,741 into an aggregate of 141,852 shares of the Company’s common stock at the conversion price of $6.00 per share, pursuant to the terms of the agreement, which conversion rate was above the closing consolidated bid price of the Company’s common stock on the date the binding agreement was entered into. (See Note 10 - Loans Payable and Note 11 - Convertible Notes Payable for more information.)
Alpha Capital Settlement
During the third quarter of 2021, the Company issued 150,000 shares of common stock and warrants to purchase 25,000 shares in connection with a settlement entered into with Alpha Capital. (see Note 11 - Convertible Notes Payable).
Common Stock Issued for Services
During the year ended December 31, 2021, the Company issued an aggregate of 317,553 shares of the Company’s common stock, respectively, as compensation to consultants, directors, and officers, with an aggregate issuance date fair value of $1,785,366, respectively, which was charged immediately to the consolidated statement of operations for the year ended December 31, 2021.
Special Voting Shares
The Special Voting Shares were issued to the former shareholders of CBR Pharma and Katexco in connection with the reorganization of 180 prior to the Business Combination. The Special Voting Shares are exchangeable by the holder for shares of the Company’s common stock and vote together as a single class with the Company’s common stockholders. Special Voting Shares are not entitled to receive any dividend of distributions.
During the year ended December 31, 2021, 1,464,545 shares were issued upon the exchange of common stock equivalents associated with the Special Voting Shares.
The following table summarizes the Special Voting Shares activity during the years ended December 31, 2021 and 2020:
| Balance, January 1, 2020 | 2,990,904 | |||
| Shares issued | - | |||
| Shares exchanged | (1,521,157 | ) | ||
| Balance, December 31, 2020 | 1,469,747 | |||
| Shares issued | - | |||
| Shares exchanged | (1,464,472 | ) | ||
| Balance, December 31, 2021 | 5,275 |
F-50
Stock Options
A summary of the option activity during the years ended December 31, 2021 and 2020 is present below:
| Weighted | Weighted | |||||||||||||||
| Average | Average | |||||||||||||||
| Number of | Exercise | Remaining | Intrinsic | |||||||||||||
| Options | Price | Term (Yrs) | Value | |||||||||||||
| Outstanding, January 1, 2020 | ||||||||||||||||
| Granted | 50,000 | 2.49 | - | - | ||||||||||||
| Exercised | - | - | - | - | ||||||||||||
| Expired | - | - | - | - | ||||||||||||
| Forfeited | - | - | - | - | ||||||||||||
| Outstanding, December 31, 2020 | 50,000 | 2.49 | 9.92 | |||||||||||||
| Granted | 2,691,000 | 4.82 | - | - | ||||||||||||
| Exercised | - | - | - | - | ||||||||||||
| Expired | - | - | - | - | ||||||||||||
| Forfeited | - | - | - | - | ||||||||||||
| Outstanding, December 31, 2021 | 2,741,000 | 4.77 | 9.41 | $ | 70,500 | |||||||||||
| Exercisable, December 31, 2021 | 927,632 | 4.39 | 9.30 | $ | 70,500 | |||||||||||
A summary of outstanding and exercisable stock options as of December 31, 2021 is presented below:
| Stock Options Outstanding | Stock Options Exercisable | |||||||||||||
| Weighted | ||||||||||||||
| Average | ||||||||||||||
| Exercise | Number of | Remaining | Number of | |||||||||||
| Price | Shares | Life in Years | Shares | |||||||||||
| $ | 2.49 | 50,000 | 8.9 | 50,000 | ||||||||||
| $ | 4.43 | 1,580,000 | 9.2 | 667,111 | ||||||||||
| $ | 7.56 | 436,000 | 9.6 | 45,417 | ||||||||||
| $ | 3.95 | 675,000 | 9.9 | 165,104 |
||||||||||
| 2,741,000 | 9.4 | 927,632 |
||||||||||||
On December 3, 2020, the Company issued ten-year options for the purchase of an aggregate of 50,000 shares of its common stock to two members of the board of directors. The options are exercisable at $2.49 per share and vest in equal monthly installments over the twelve months following the grant date. The grant date value of $93,575 was estimated using the Black Scholes valuation method with the following assumptions used:
| Risk free interest rate | 0.4 | % | ||
| Expected term (years) | 5.27 | |||
| Expected volatility | 100 | % | ||
| Expected dividends | 0 | % |
On February 26, 2021, the Company issued ten-year options to purchase an aggregate of 1,580,000 shares of the Company’s common stock to two officers of the Company, pursuant to the 2020 Omnibus Incentive Plan. The options have an exercise price of $4.43 per share and vest at the rate of 20% on the date of grant and the remaining 80% on a monthly basis thereafter over the following 36 months. The options had a grant date fair value of $4,810,527, which will be recognized over the vesting term.
F-51
On August 4, 2021, the Company granted ten-year options for the purchase of an aggregate of 436,000 shares of common stock at an exercise price of $7.56 per share, to six independent directors of the Company, pursuant to the 2020 Omnibus Incentive Plan. The options had an aggregate grant date value of $2,180,375, and vest monthly over four years.
On December 8, 2021, the Company granted ten-year options for the purchase of an aggregate of 675,000 shares of common stock at an exercise price of $3.95 per share to six officers of the Company, pursuant to the 2020 Omnibus Incentive Plan. The options had an aggregate grant date value of $2,077,953 and vest at various periods over four years.
The assumptions used in the Black-Scholes valuation method were as follows:
| Risk free interest rate | 0.75% - 1.36% | |
| Expected term (years) | 5.61 - 6.01 | |
| Expected volatility | 84% - 98.5% | |
| Expected dividends | 0% |
The Company recognized stock-based compensation expense of $2,852,309 and $7,798 for the years ended December 31, 2021 and 2020, respectively, related to the amortization of stock options. The expense is included within general and administrative expenses or research and development expenses on the consolidated statements of operations. As of December 31, 2021, there was $6,302,356 of unrecognized stock-based compensation expense that will be recognized over the weighted average remaining vesting period of 3.04 years.
NOTE 14 — INCOME TAXES
The Company is subject to federal and state/provincial income taxes in the United States, Canada, and the United Kingdom and each legal entity files on a non- consolidated basis. The benefit of the pre-reorganization net operating losses of 180 LP were passed through to its owners.
The losses before income taxes consist of the following domestic and international components:
| For the Years Ended | ||||||||
| December 31, | ||||||||
| 2021 | 2020 | |||||||
| Domestic | $ | (15,078,170 | ) | $ | (8,635,341 | ) | ||
| International | (5,269,682 | ) | (2,269,144 | ) | ||||
| $ | (20,347,852 | ) | $ | (10,904,485 | ) | |||
The provision for income taxes consists of the following benefits (provisions):
| For the Years Ended | ||||||||
| December 31, | ||||||||
| 2021 | 2020 | |||||||
| Deferred tax benefits: | ||||||||
| Domestic: | ||||||||
| Federal | $ | 1,503,577 | $ | 1,289,907 | ||||
| State | 499,136 | 427,689 | ||||||
| International | 547,944 | 541,614 | ||||||
| 2,550,657 | 2,259,210 | |||||||
| Change in valuation allowance | (2,527,453 | ) | (2,238,783 | ) | ||||
| Net income tax benefit | $ | 23,204 | $ | 20,427 | ||||
F-52
Certain deferred tax liabilities are denominated in currencies other than the US dollar and are subject to foreign currency translation adjustments. The provision for income taxes differs from the United States Federal statutory rate as follows:
| For the Years Ended | ||||||||
| December 31, | ||||||||
| 2021 | 2020 | |||||||
| US Federal statutory rate | 21.0 | % | 21.0 | % | ||||
| Difference between domestic and foreign federal rates | (0.5 | )% | (0.6 | )% | ||||
| State and provincial taxes, net of federal benefits | 5.2 | % | 6.0 | % | ||||
| Permanent differences: | ||||||||
| Stock-based compensation | (5.8 | )% | (0.8 | )% | ||||
| Change in the fair value of derivatives and accrued issuable equity | (6.4 | )% | (4.6 | )% | ||||
| Loss on extinguishment | - | (1.0 | )% | |||||
| Other | (0.8 | )% | 0.7 | % | ||||
| Change in valuation allowance | (12.4 | )% | (20.5 | )% | ||||
| Effective income tax rate | 0.3 | % | 0.2 | % | ||||
Deferred tax assets and liabilities consist of the following:
| As of December 31, | ||||||||
| 2021 | 2020 | |||||||
| Deferred tax assets: | ||||||||
| Net operating loss carryforwards | $ | 9,395,986 | $ | 6,352,809 | ||||
| Organizational costs deferred for tax purposes | - | 3,068,651 | ||||||
| Accrued compensation not currently deductible | 169,222 | 224,931 | ||||||
| Accrued interest | 146,636 | - | ||||||
| Other | (1 | ) | 62,829 | |||||
| 9,711,843 | 9,709,220 | |||||||
| Deferred tax liabilities: | ||||||||
| Difference between book and tax basis related to: | ||||||||
| Technology license | (375,671 | ) | (404,507 | ) | ||||
| Acquired in-process research and development | (3,267,854 | ) | (3,242,750 | ) | ||||
| Other | (639,726 | ) | (21,072 | ) | ||||
| (4,283,251 | ) | (3,668,329 | ) | |||||
| Deferred tax assets and liabilities | 5,428,592 | 6,040,891 | ||||||
| Valuation allowance | (9,072,118 | ) | (9,709,220 | ) | ||||
| Deferred tax assets and liabilities, net | $ | (3,643,526 | ) | $ | (3,668,329 | ) | ||
F-53
The change in the valuation reserve for deferred tax assets consists of the following:
| For the Years Ended | ||||||||
| December 31, | ||||||||
| 2021 | 2020 | |||||||
| Beginning of period | $ | (9,709,220 | ) | $ | (4,979,276 | ) | ||
| Allowance established in connection with the recording of deferred tax assets acquired resulting from the following transactions: | ||||||||
| - Business combination in 2020 described in Note 5 | - | (2,491,161 | ) | |||||
| Change in valuation pursuant to the tax provision | (2,527,453 | ) | (2,238,783 | ) | ||||
| True-up to a prior year’s tax return | 3,164,555 | - | ||||||
| End of period | $ | (9,072,118 | ) | $ | (9,709,220 | ) | ||
As of December 31, 2021, the Company had net operating loss (“NOL”) carryforwards that may be available to offset future taxable income in various jurisdictions as follows:
| ● | Approximately $13,330,000 of domestic federal and $7,359,000 of state NOLs. The federal NOLs have no expiration date and the state NOLs will begin to expire in 2039; |
| ● | Approximately $ 8,589,000 each of Canadian federal and provincial NOLs. Those NOLs will begin to expire in 2038; and |
| ● | Approximately $6,791,000 of United Kingdom federal NOLs. Those NOLs have no expiration date. |
The utilization of the domestic NOLs to offset future taxable income may be subject to annual limitations under Section 382 of the Internal Revenue Code and similar state statutes as a result of ownership changes.
On November 6, 2020, we acquired net deferred tax assets of $2,491,161, against which there is a full valuation allowance.
The Company has assessed the likelihood that deferred tax assets will be realized in accordance with the provisions of ASC 740 Income Taxes (“ASC 740”). ASC 740 requires that such a review considers all available positive and negative evidence, including the scheduled reversal of deferred tax liabilities, projected future taxable income, and tax planning strategies. ASC 740 requires that a valuation allowance be established when it is “more likely than not” that all, or a portion of, deferred tax assets will not be realized. After the performance of such reviews as of December 31, 2021 and 2020, management believes that uncertainty exists with respect to future realization of its deferred tax assets and has, therefore, established a full valuation allowance as of those dates. Thus, the Company established a valuation reserve of $2,491,161 in connection with the net deferred tax assets acquired in connection with the Business Combination described in Note 5 during the year ended December 31, 2020. Additionally, the Company recorded increases in the valuation allowance of $2,527,453 and $2,238,783 in connection with the tax provisions for the years ended December 31, 2021 and 2020, respectively.
Management has evaluated and concluded that there were no material uncertain tax positions requiring recognition in the Company’s consolidated financial statements as of December 31, 2021 and 2020. The Company does not expect any significant changes in its unrecognized tax benefits within twelve months of the reporting date.
No tax audits were commenced or were in process during the years ended December 31, 2021 and 2020 nor were any tax related interest or penalties incurred during those periods. The Company’s tax returns filed in the United States, Canada, and the United Kingdom since inception remain subject to examination, with the exception of the tax returns filed for the 180 LP pass-through entity whose tax returns remain subject to examination beginning with the 2018 tax return.
F-54
NOTE 15 - RELATED PARTIES
Due from Related Parties
Due from related parties of $300,000 as of December 31, 2020 consisted of a receivable due from a research and development company that has shared officers and directors. Management now believes that the receivable is not collectible, and the receivable is fully reserved as of December 31, 2021.
Accounts Payable - Related Parties
Accounts payable - related parties was $0 as of December 31, 2021. Accounts payable - related parties was $215,495 as of December 31, 2020 and consists of $196,377 for professional services provided by the Company’s directors and $19,118 for accounting fees for services provided by a former director and his company.
Accrued Expenses - Related Parties
Accrued expenses - related parties was $18,370 as of December 31, 2021 and consists of interest accrued on loans and convertible notes due to certain officers and directors of the Company. Accrued expenses - related parties of $454,951 as of December 31, 2020, consists of $124,833 of interest accrued on loans and convertible notes due to certain officers and directors of the Company and $330,118 of accrued professional fees for services provided by certain directors of the Company.
Loans Payable - Related Parties
Loans payable - related parties consists of $81,277 and $513,082 as of December 31, 2021 and 2020, respectively. See Note 10 - Loans Payable for more information.
Convertible Notes Payable - Related Parties
Convertible notes payable - related parties of $0 and $270,000 as of December 31, 2021 and 2020, respectively, represents the principal balance of convertible notes owed to certain officers and directors of the Company. See Note 11 - Convertible Notes Payable for more information.
Research and Development Expenses - Related Parties
Research and Development Expenses – Related Parties of $2,947,536 and $75,633 during the years ended December 31, 2021 and 2020, respectively, is related to consulting and professional fees paid to current or former officers, directors or greater than 10% investors, or affiliates thereof.
General and Administrative Expenses - Related Parties
General and Administrative Expenses – Related Parties during the years ended December 31, 2021 and 2020, were $462,580 and $185,848, respectively. Of the expenses incurred during 2021, approximately $338,000 represents bad debt expense incurred in connection with a receivable from related parties, and approximately $124,000 represents professional fees paid to current or former officers, directors or greater than 10% investors, or affiliates thereof. The expenses incurred during 2020 relate to professional fees paid to current or former officers, directors or greater than 10% investors, or affiliates thereof.
Other Income - Related Parties
Other Income – Related Parties during the years ended December 31, 2021 and 2020, were $0 and $240,000, respectively. During 2020, the Company recorded $240,000 of other income related to a one-year research and development agreement with a company who shares common officers and directors with the Company.
F-55
Interest Expense - Related Parties
During the year ended December 31, 2021, the Company recorded $50,255 of interest expense – related parties, of which $11,380 related to the convertible notes with officers and directors of the Company and $38,875 related to interest expense on loans with officers, directors and a greater than 10% investor of the Company.
During the year ended December 31, 2020, the Company recorded $84,550 of interest expense – related parties, of which $48,591 related to the convertible notes with officers and directors of the Company and $33,798 related to interest expense on loans with officers, directors and a greater than 10% investor of the Company, and $2,161 was incurred prior to the Reorganization in connection with a Katexco loan payable to CBR Pharma.
NOTE 16 - SUBSEQUENT EVENTS
The Company has evaluated events and transactions subsequent to December 31, 2021 through the date the financial statements were issued. Except for the following, there are no subsequent events identified that would require disclosure in the financial statements.
Board of Directors Compensation – Fourth Quarter 2021
On January 17, 2022, the Company paid $88,125 in cash and shares of common stock to the Board of Directors for compensation for the fourth quarter of 2021.
ACORN Consulting Agreement
On February 22, 2022, the Company entered into a consulting agreement with ACORN, a professional relations and consulting firm. ACORN will be engaged to create a full market awareness program, drive long-term investors to the Company and expand the Company’s investor base. The Company will pay ACORN $10,000 in cash and $45,000 in shares of common stock during an initial six-month period, $10,000 in cash and $22,500 in shares of common stock during a second period of three months and $10,000 in cash and $22,500 in shares of common stock during a final period of three months. These compensation shares will be fully vested, earned, authorized and paid upon issuance.
MDM Worldwide Solutions, Inc. Consulting Agreement
On March 15, 2022, the Company entered into a consulting agreement with MDM Worldwide Solutions, Inc. (“MDM”). MDM will be engaged to provide the Company with strategic communication-based advisory and consulting services for the purpose of targeted outreach and engagement with the inflammatory disease communities online. The Company will pay MDM $12,500 in cash per month and a one-time 20,000 shares of restricted common stock, which will be deemed earned upon execution of the contract. The initial term of the contract is six months.
Amendment to the Fourth Oxford Agreement
On March 22, 2022, CannU.K. entered into an amendment to the Fourth Oxford Agreement, which was originally signed on September 21, 2020, to extend the research period to December 31, 2023, at no additional cost to CannU.K.
Update – meeting with FDA regarding treatment of Dupuytren’s Contracture
In March 2022, the Company received notice that the FDA had declined to grant a meeting with the Company regarding its preliminary discussion, Type C meeting for its treatment of early stage Dupuytren’s Contracture, until more information about the manufacturer of the product is provided.
The Company remains committed to the development of its early stage Dupuytren’s Contracture treatment and upon advice of its regulatory consultants plans to further correspond with the FDA with the goal of having a Type C meeting with the FDA; provided no assurances can be given that such meeting will be agreed to by the FDA or the timing of such meeting.
F-56